Molecular dynamics data will be essential for the next generation of ML protein models
post by Abhishaike Mahajan (abhishaike-mahajan) · 2024-08-26T14:50:23.790Z · LW · GW · 0 commentsThis is a link post for https://www.owlposting.com/p/an-argument-for-integrating-molecular
Contents
Introduction The arguments Biology models don’t understand physics Models that learn from physics are better models We are running out of structural and sequence data TLDR Building better datasets What the future looks like Conclusion None No comments

Introduction
I’ve been pondering the thesis behind this post for a few months now, figuring out how to approach it. In my head, it feels plainly obvious, of course we should use molecular dynamics (MD) to help further train proteomics models. But it’s a good exercise to motivate the whole thing from a first-ish-principles place. Upon writing this, I realized a lot of my initial thoughts about the subject were misguided or misinterpreted. Hopefully this synthesis helps someone understand the role MD will play in the future of proteomics ML.
In this post, I’ll sketch out three reasons why I believe MD will be fundamental in the next generation of proteomics models, each one building off each other. We’ll then end with a brief thought on what will be necessary to produce this next generation of models. We’ll also quickly point out one recently released paper that I would bet is an early precursor of what’s to come.
Quick note: we won’t discuss things stuff like neural-network potentials in this essay, which are set up to change MD itself (and maybe discussed in a future essay). Instead, we’ll focus entirely on how even the current era of MD is sufficient to dramatically benefit proteomics models.
The arguments
Biology models don’t understand physics
Protein folding models, such as AlphaFold2 (and, recently, AlphaFold3) represent the clearest success of ML applied in the life sciences. In many ways, the single-chain protein structure prediction problem is largely solved, though a long tail of edge cases exists (and will likely continue to exist for a while).
But models like Alphafold2 (AF2) do not work by simulating the physics of a protein. No ML-based folding model seems to, not OmegaFold, not ESM2, none of them. When AF2 first came out, it was likely hypothesized that it had somehow learned a fuzzy notion of physics from end-state structures alone. This was quickly called into suspicion by a 2022 paper titled ‘Current structure predictors are not learning the physics of protein folding’, which found that ‘folding trajectories’ produced by Alphafold2 (the details of which could be found in section 1.14 here) do not recapitulate real folding dynamics at all. This was reaffirmed in a 2023 paper titled ‘Using AlphaFold to predict the impact of single mutations on protein stability and function’, which studied whether Alphafold2 predicted confidence correlated with experimental stability for point mutations. They didn’t! The class of structure prediction models most likely to have learnt a strong notion of biophysics — protein language models, as they do not require MSA’s — also have been found to work via implicitly learned coevolutionary information.
This all said, it’s worth mentioning that there is an argument that these models do have some vague notion of physics: they work decently for proteins with little-to-no MSA information. The strongly titled paper ‘Language models generalize beyond natural proteins‘ found exactly this. But they do not claim that this means anything about whether these models have learned physics, but rather that a ‘deep grammar’ underlies all functional proteins, which is perhaps ruled by physics, but does not require understanding physics itself to derive:
This generalization points to a deeper structure underlying natural sequences, and to the existence of a deep grammar that is learnable by a language model. Our results suggest that the vast extent of protein sequences created through evolution contains an image of biological structure and function that reveals design patterns that apply across proteins, that can be learned and recombined by a fully sequence based model. The generalization beyond natural proteins does not necessarily indicate that language models are learning a physical energy. Language models may still be learning patterns, rather than the physical energy, but speculatively, in the limit of infinite sequence data, these patterns might approximate the physical energy. At a minimum the language model must have developed an understanding of the global coherence of a protein connecting the sequence and folded structure.
So, it is still unlikely that these models understand physics — there simply is some universal pattern underlying most proteins in existence. But this universal pattern only seems to take you so far with the current era of models, there are still very likely a massive number of failure modes.
Okay, so, folding models don’t understand physics. Why is this a problem? Why do we care? Let’s say we can magically create a version of Alphafold that intuitively gets electrostatic energy on some abstract level. Why does this help us in any capacity beyond being theoretically interesting?
That leads well into our next point!
Models that learn from physics are better models
There is, I think, some hesitation in combining physics and ML. After all, it’s a strong prior to place on a model, and priors are increasingly out of vogue in the field. Models like Physics-Informed Neural Networks (PINN), which force the model to have an inductive bias towards satisfying user-provided physical laws, have been relatively unpopular (though there is increasingly a resurgence of them). The claim there is that supervised learning from data alone is extremely inefficient for problems that are inherently bound by physics, so adding in hard constraints to the network outputs should help with extrapolation outside the training dataset.
Perhaps the future is indeed a PINN that have grounded physical laws baked into it. But maybe we could try something even simpler. Could we simply pluck out physics-based features from a molecular dynamics simulation (such as free energy calculations), throw that into a model, and observe any increases in accuracy?
Surprisingly, yes!
‘Incorporating physics to overcome data scarcity in predictive modeling of protein function’ did exactly this, deriving physically-derived features like the free-energy impact of single-point mutations —- along with more dynamical physical properties (measured every nanosecond) such as solvent-accessible surface area and RMSF — and combining them with normal amino-acid derived biochemical features.
These features were used to predict the impact of single mutations on gating voltage produced by the BK channel protein. Using physics-based features led to a large improvement:
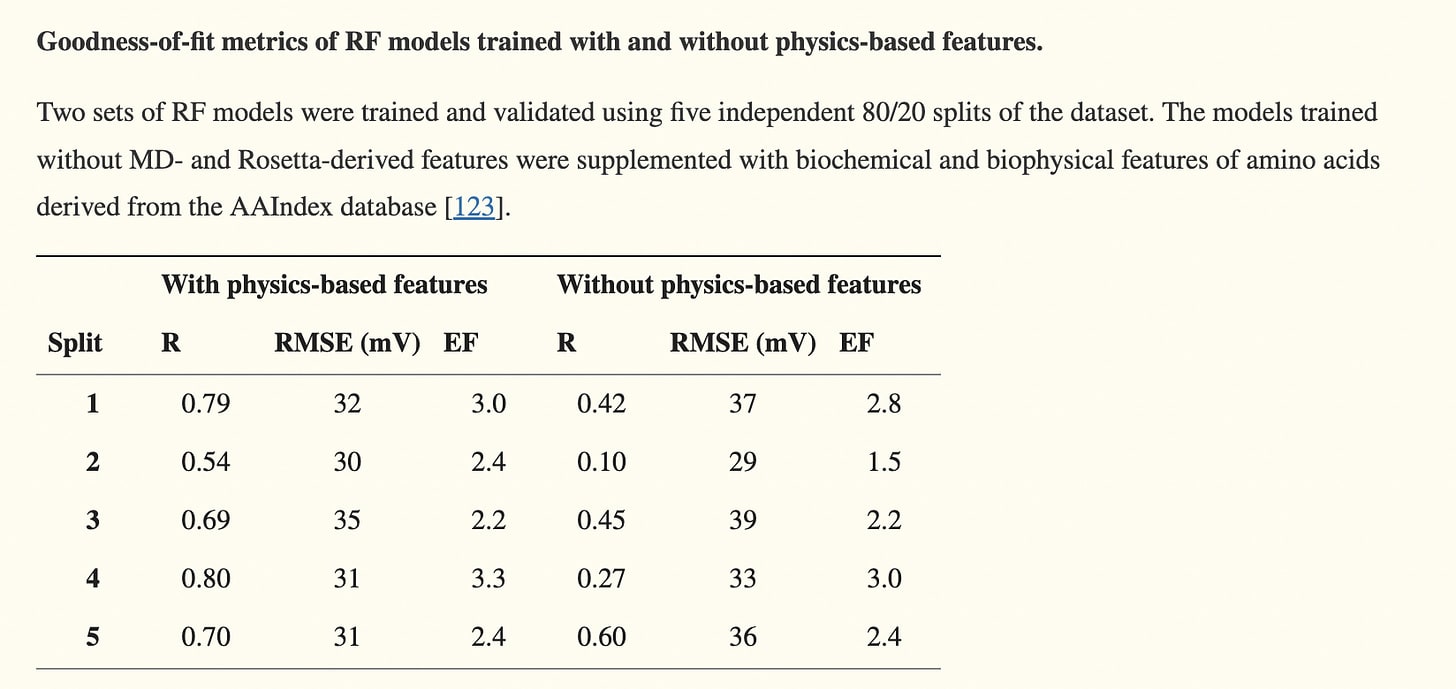
But MD trajectories are incredibly difficult to calculate, requiring vast amounts of computational resources/time for even small sets of proteins. As we’ll see later, the datasets in this space are still extremely small.
Could we instead rely on a snapshot of potential energy and structural information (also known as ‘energetics’)? This is relatively simple to derive, since you aren’t actually running an MD trajectory, just performing quick calculation of a pre-existing structure. Perhaps this alone gives us enough information for a model to understand physics?
Somewhat, but there are huge caveats that make it largely useless. Another paper titled ‘Learning from physics-based features improves protein property prediction’ investigated it. They produced the following ‘energetic’ features per protein over either 5 MD samples or 1 MD sample.

They compared these energetic features to a typical one-hot-encoding + structure feature representation network trained to predict the outcome of interest (Baseline), along with the same network having first been pre-trained on several thousand other structures first (Pretrain). Specifically pay attention the bottom table for GB1 fitness.
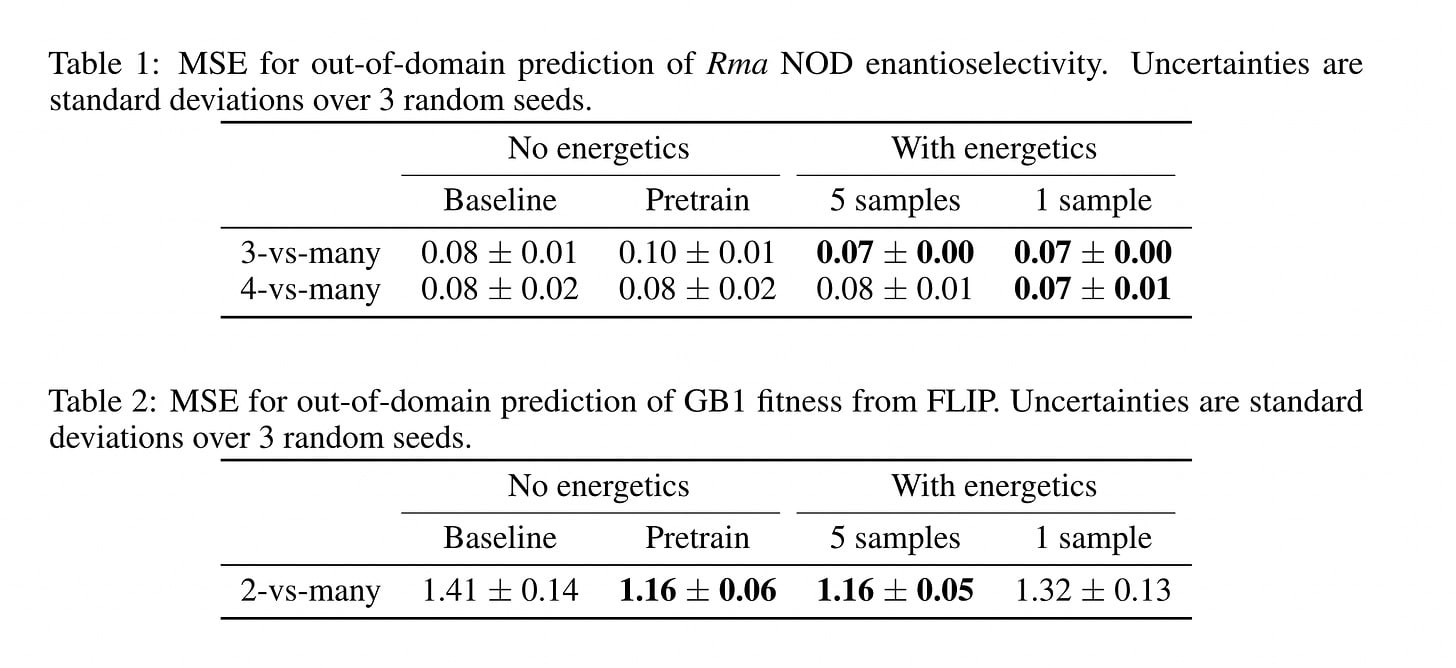
While energetic features do seem to improve performance, they only seem to improve performance up to the point of a pre-trained model! More plainly, energetics and sequences teach models the same thing. Another, much more recent, paper titled ‘Biophysics-based protein language models for protein engineering’ found nearly identical results, using only energetics features again. This method is useful only for problems with low-N datasets.
So, energetic features, which are the main ones we’re able to scale, are largely insufficient in high-N settings.
But these results may still raise a seed of doubt in our minds. How do we know dynamics-based information isn’t already encoded in models such as AlphaFold3? And, even if it hasn’t, couldn’t we get that information from scaling on sequence/structure alone? The information contained within such dynamic’s features should be derivable from sequence/structure alone, right? If that was the case for energetics features, why do we expect it to be any different for dynamics features?
A recent paper on AlphaFold3 sheds some light here. The paper, titled AlphaFold3, a secret sauce for predicting mutational effects on protein-protein interactions, finds two interesting things of note:
- MD is still superior to AF3 in predicting impact of single-mutations (a decent proxy for being able to understand physics), implying that physics-based information may still be useful to AF3
- AF3 gets far closer to MD-based results than any other model
So, AlphaFold3, which has only ever seen structures, is clearly implicitly learning the potential energy surface of any given protein. It’s not all the way there yet, MD still seems to be strictly better at this task, but the exact role of MD is uncertain given these results. Why can’t we just scale AlphaFold3 even further, with more structures, and avoid the hassle of having to work with MD? Whatever physics information is useful will be learned by the model — given enough datapoints — no need to directly feed in physics-based information.
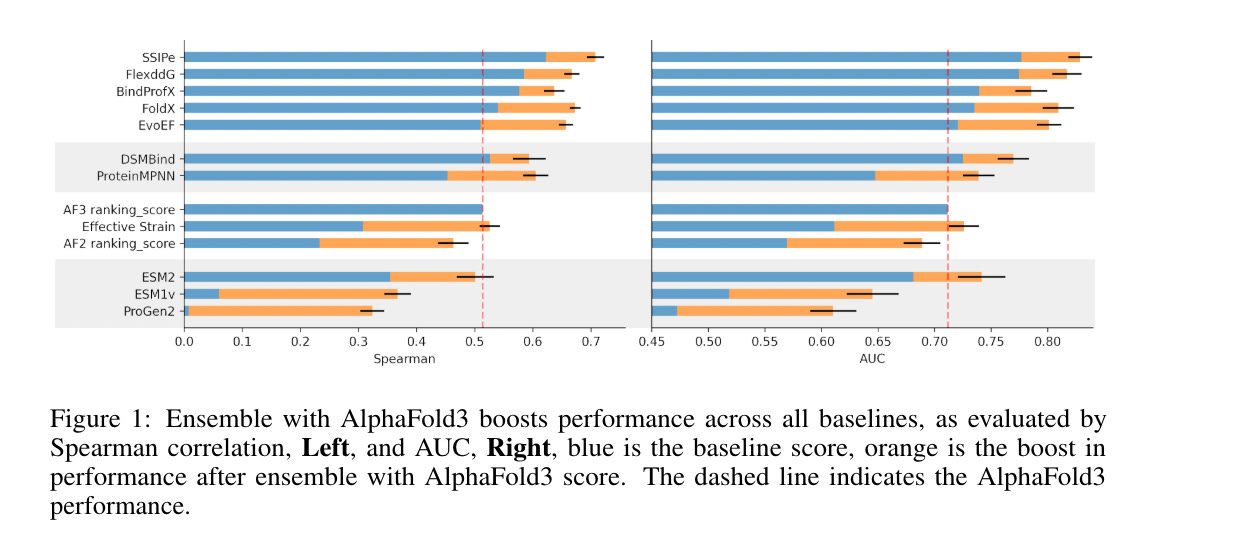
This is a fair point. We could make the argument that the physics information derivable from MD is really, really hard to get from structure/sequence alone, that this single-mutation challenge may uniquely fail for evolutionarily distinct proteins in AF3 (whereas MD will still perform well), and so on. But it’d be hard to defend any of those statements, bitter lesson and all.
So, why do we need MD?
We are running out of structural and sequence data
Here’s why this post is an ‘argument’. Up to this point, I haven’t really said anything that is completely unsubstantiated. But now I will: the biology-ML field is running out of useful sequence and structural data to train models.
So even if we could fully derive physics-based information from sequences/structures, it is likely we don’t have enough datapoints for that.
There is no real way for me to prove this. But there are signs!
The recent AlphaFold3 actually heavily points to this direction. It seems to derive most of its improvements from architecture changes, expanded MSA databases, distillation, and (most notably) transfer learning from the addition of new biomolecules to model. But there were no sizable changes/additions to its input sequence/structure training dataset, it still mainly relies entirely on the same PDB. How many experimentally determined structures could really be left to train on?

From here.
Okay, so, structural data is likely a bit limited due to cost of acquisition, but what about sequences? Being able to mass-collect metagenomic data means we surely aren’t limited in that realm, right?
xTrimoPGLM in early 2024 (or XT100B), which was really pushing the sequence-only scale hypothesis, disproves this at least a little. It used UniRef90, alongside several massive metagenomic databases (BFD, MGnify, and several others), as training data, resulting in around 940M unique sequences and 20B unique tokens. Despite the trained model having 100B parameters and being trained on 1T tokens in total — which is both far larger than other models and has a nice Chinchilla-scaling ratio — the results aren’t anything extraordinary. While it beats out most other models across a wide range of tasks, it is, for the most part, a modest increase in performance.
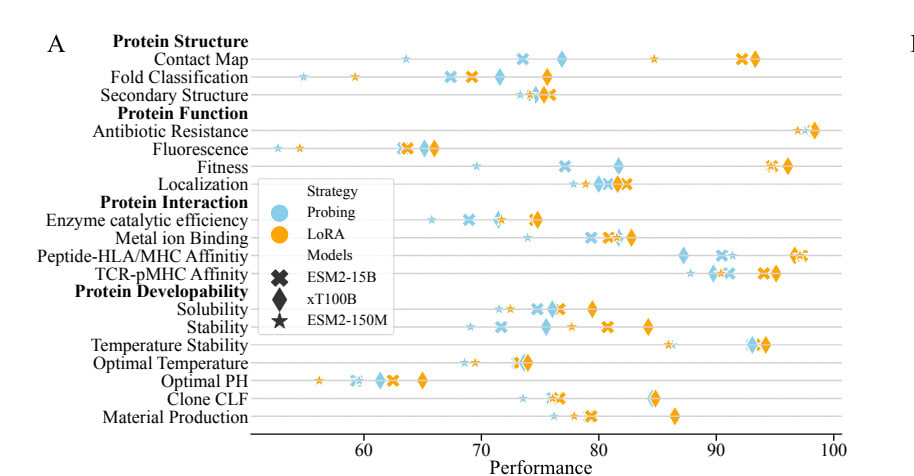
From here.
Again, I can’t prove that these diminishing returns will absolutely be the case in the upcoming future. Maybe I’m wrong!
Maybe proteomics inheriting the NLP pre-training tasks (autoregressive or masked modeling) is insufficient, and future, more clever pre-training tasks will help make the existing structure/sequence data more useful. Maybe better architectures will pop up. Maybe the inclusion of NMR or cryo-EM structures, small as they are, will still help an immense amount. Maybe most metagenomic data is still largely in-distribution, and companies like Basecamp Research will be able to find more O.O.D data to train our models with.
But I’m a little skeptical.
TLDR
TLDR: current protein models don’t understand physics, physics is useful for understanding biomolecules, MD can (probably) teach models aspects of physics, and it is unlikely that we will ever have enough proxies-of-physics (sequences/structures) for models to implicitly gain that understanding.
Now what?
Building better datasets
What is the next step? The most important one is that we need more standardized, large MD datasets, especially focused on larger biomolecules. The vast majority of existing ones, though large in size (100k-1M~ datapoints), primarily focus on purely small molecule modeling or purely peptide modeling. These are both important in their own right! But larger ones will almost certainly be necessary.
There are already several here for the classical mechanics side, such as ATLAS, which includes 1.5k~ all-atom MD simulations of 38+ residue proteins. For protein-ligands, there is PLAS-20k, which, as the name implies, contains 20k protein-ligand pairs and nearly 100k~ total trajectories. For antibody-antigen docking, there is ThermoPCD, which only have 50 total complexes, but provides the trajectories at several different temperatures.
Quantum datasets will likely play an important role too, given that classical mechanics simulations are often inaccurate. An example of this is MISATO, which is quantum-ish (quantum force fields are only used on the ligand), containing 20k trajectories for protein-ligand pairs. But generally, this area of datasets lags far behind the state-of-the-art in classical mechanics.
It’s very much early days, many of the MD dataset curation papers in the field have only been published in the last 1-2 years!
What the future looks like
Let’s say we build this massive MD dataset. How exactly do we feed it into models?
Basic options work well, as we’ve discussed above. While many papers have used more complex thermodynamics features, such as delta free energy calculations, as input to their models, that isn’t strictly necessary to capture a sense of dynamics. One paper, Assessment of molecular dynamics time series descriptors in protein-ligand affinity prediction, used basic tsfresh features extracted from the MD trajectory as features and still found minor performance improvements compared to crystal-structure-only features. Of course, this may be falling prey to the same issue as before, where this advantage will disappear upon scaling the sequence dataset size. Thermodynamic features, such as free energy calculations, likely have the strongest signal (given that even AF3 couldn’t outperform it) but seem challenging to calculate at scale.
To truly take advantage of trajectories in a way amenable to 1M+ trajectories, models will likely need to operate directly on the trajectories themselves rather than represent them via hand-crafted features. As seems to be the case for all ML ideas, this was done just a few months ago (Feb 2024) in a reasonably well-known model: AlphaFlow. It is first trained on the PDB, and then further trained on the aforementioned ATLAS datasets with a flow-matching loss. At run-time, it can create a simulated 300 nanosecond trajectories given sequence + MSA alone.
People familiar with the paper may be surprised I’m mentioning it here at all! It isn’t meant to improve performance on tasks such as structure prediction, but rather just be a faster and more accurate way to gather up protein conformations given a sequence. In this respect it does fine, there is an interesting Twitter thread that asks for MD expert opinions on the paper. Generally positive but there’s a lot of room for improvement.
But the far more interesting part is what AlphaFlow has internally learned about physics and how these transfer to downstream tasks. On the surface, it clearly understands protein flexibility decently, being able to recapitulate true protein dynamics far better than AF2 methods such as MSA subsampling. But how this transfers to new emergent capabilities is still unknown. Keep in mind, it is unlikely that AlphaFlow alone will be a step change in any capacity! The dataset it uses for MD training, ATLAS, is still quite small, over relatively short time spans, and is based on only classical mechanics. But AlphaFlow represents the first (in my opinion) public release of what the next generation of protein models will look like: a synthesis between sequences/structures and molecular dynamics trajectories.
Conclusion
The field of proteomics is at an inflection point. The current generation of models, while impressive, are fundamentally limited by their lack of understanding of the underlying physics governing protein behavior. This is not a failing of the models themselves, but rather a reflection of the data they are trained on; sequences and static structures alone are insufficient to capture the complex dynamics of proteins in their native environments, at least at the current dataset sizes we currently have. While Alphafold3 does seem to be poking at an understanding of these dynamics from structure alone, I am unsure what non-MD-tricks are left to really close the gap — and ideally go far past MD alone.
The future is incorporating MD data into the training process! Of course, this is easier said than done. MD simulations are extraordinarily computationally intensive, and generating large-scale datasets will require significant resources. I am unsure who will spearhead this effort, the state of large-scale MD simulations is really only in the realm of supercomputers. Perhaps DESRES, Isomorphic Labs, or someone else will be the first here, akin to the OpenAI of the biology foundation model world.
There is a mild concern I have here. The early days of biology-ML were heavily assisted by the ML culture it bathed in; lots of transparency, data sharing, code sharing, and so on. But there will reach a point where these models become fundamentally valuable in the ultimate goal of actually delivering a drug. And it’s hard to overstate how immensely profitable a drug can be; the canonical blockbuster drug Humira brings in tens of billions a year and has done so for 10+ years. When this time comes, we may see an end to the radical transparency of the field, as previously transparent institutions feverishly protect the secrets behind a potential money printer. This alone isn’t the end of the world; NLP is currently going through something similar.
But if something as computationally intensive, difficult to create, and esoteric as MD becomes foundational to the next generation of models, as opposed to the open-sourced PDB and sequence databases, an open-source response to Alphafold4, Alphafold5, and so on may become impossible. It is unlikely that models like AlphaFold3 are at that level yet, but the early walling-off of AlphaFold3 (despite it soon being released for academic use), is likely a sign of what’s to come. It is deeply unfortunate that Meta fired their protein AI team, they would be my first hope for an open-source response to Isomorphic Labs models, given all their work in Llama3. The OpenFold Consortium may end up being the primary leader here, as they replicated Alphafold2, but time will tell. Remember, open source is important for everybody, reducing technical barriers for curious/enthusiastic people helps both for-profit and non-profit entities alike, it’d be very bad for the field if we saw a shuttering of publicly released models.
At the same time, this may end up being a non-issue. I can very much see a world in which neural-network potentials dramatically speed up the MD data acquisition process. And another world in which MD is valuable, but one needn’t get millions upon millions of trajectories to learn something useful, merely a few thousand may be enough to get 80% of the predictive benefit. Again, time will tell.
Either way, I feel strongly that MD and ML will be strongly intertwined in the years to come. Very excited to see how things progress from here on out!
0 comments
Comments sorted by top scores.