Better antibodies by engineering targets, not engineering antibodies (Nabla Bio)
post by Abhishaike Mahajan (abhishaike-mahajan) · 2025-01-13T15:05:35.261Z · LW · GW · 0 commentsThis is a link post for https://www.owlposting.com/p/better-antibodies-by-engineering
Contents
Introduction The pain of multi-pass membrane proteins (MPMP) Computational design of MPMP proxies Joint Atomic Modeling Conclusion None No comments

Note: Thank you to Surge Biswas (founder of Nabla Bio) for comments on this draft and and Dylan Reid (an investor into Nabla) for various antibody discussions! Also, thank you to Martin Pacesa for adding some insight on a paper of his I discuss here (his comments are included).
Introduction
Antibody design startups are singlehandedly the most common archetype of bio-ML startup out there. It’s understandable why — antibodies are derisked modalities, CDR loops driving antibody efficacy makes the whole structure more amenable to ML, and there’s a fair bit of pre-existing data there. But, because it’s also the most common form of company, it’s difficult to really differentiate one over the other.
If you squint, you could make out some vague distinguishing characteristics. Bighat Biosciences does a lot of multi-property optimization, Absci and Prescient have the strongest external research presence, and so on. But there is a vibe of uniformity. It’s nobody’s fault, that’s just the nature of any subfield that has a huge amount of money flowing into it; everyone quickly optimizes. And, unfortunately for those of us who enjoy some heterogeneity, most everyone arrives at the same local minima.
Because of that, I’ve never really wanted to write about any antibody company in particular. None of them felt like they had a sufficiently interesting story. All fine companies in their own right, but they all tell the same tale: great scientists, great high-throughput assays, great machine-learning, and so on.
This brings us to the topic of this essay: Nabla Bio.
As may be expected, from the earlier sections of this piece, they are an antibody design startup. Founded by Surge Biswas, an ex-Church Lab student who worked with ML-guided protein modeling during his PhD, and Frances Anastassacos, they were launched in 2021 and currently sit at 15 employees. Nabla, on the surface, looks materially indistinguishable from most other antibody design companies. And, in many ways, they are! If you visit their website, you’ll immediately see references to barcoding, parallel screening, and machine learning. It feels like much of the same story as everyone else.
It took me until a few months later for me to realize that Nabla has an interesting difference. Because Nabla is not only an antibody engineering company. They may say they are, and their partnerships may only include antibodies, but I don’t consider that their defining archetype alone.
What they really are, alongside antibodies, is a target engineering company. And that is why, amongst companies they are often compared to, I find them uniquely curious. This target engineering thesis they are pursuing is the subject of today's essay.
But before we talk about them, we first need to talk about targets.
The pain of multi-pass membrane proteins (MPMP)
What makes for a good drug target? Of course, the most critical and obvious factor is therapeutic potential. Will modulating this target actually help treat or cure the disease? This is the fundamental requirement that drives target selection. However, there's often a disconnect between knowing a target is therapeutically valuable and being able to successfully develop drugs against it.
So, given a set of drug targets that all are known to be related to a disease of interest, how do you pick amongst them? There’s a lot of ways you could go about filtering them, including market interest, how much it’s been clinically derisked, and so on. But an often used method for selecting a target is how easy it is to work with.
What are the phenomena of targets that are easy to work with? Here are three that come to mind:
- Stable, because having a protein that maintains a consistent structural and functional identity across time and varying conditions makes in-vitro testing with it far easier. When you're running high-throughput screens or binding assays, you’d ideally like for your target protein to not unfold, aggregate, or adopt wildly different conformations between experiments. Stability, in the end, really translates to predictability.
- Well-characterized, because a protein we understand the behavior of is a protein that we can exploit. Binding pockets, conformational changes, and interaction partners of a target can all be helpful things to keep in mind throughout the life cycle of a therapeutic designed to interact with it.
- Amenable to being bound to by your therapeutic molecule of choice, because how else would a therapeutic interact with it?
MPMPs fail spectacularly on almost all these counts.
MPMP’s are amphipathic proteins, meaning having both hydrophilic and hydrophobic components. This should make intuitive sense. In their native context, their hydrophobic transmembrane segments are buried in the cell membrane's lipid bilayer, while their hydrophilic regions extend into the aqueous environments on either side. Importantly, MPMP’s are not only embedded into the cell membrane, but weave through it multiple times. If it wasn’t obvious, that’s what the ‘multi-pass’ bit of the acronym stands for. This means we have alternating, repeating stretches of hydrophobic-hydrophilic residues.
This makes MPMP’s awful to work with.
A fair number of other proteins in the human proteome are water-soluble — adapted to exist in the (mostly) aqueous environment of the cell's cytoplasm or extracellular space. These are fantastic to work with! You can extract them, purify them, and work with them while maintaining their native structure. This has a lot of second-order value: it’s easier to run experiments with them while having consistent results, it’s easier to characterize their structure using experimental determination techniques, and it’s way cheaper to get them ready for whatever assay you want to run with them.
MPMP’s have no such advantages.
When isolated out of their membrane environment to study individually, it’s a struggle to recapitulate their normal behavior. Their hydrophobic segments, normally protected by the membrane's lipid environment, become exposed to an aqueous environment. This is about as stable as you might expect: these proteins tend to rapidly misfold, aggregate with one another, or completely fall apart. If you want to run a high throughput screen, it’ll be a constant challenge to get consistent protein conformations across your assay conditions. If you want structural data, keeping the protein stable long enough to even attempt crystallization or cryo-EM will be enormously difficult. And even if you manage all of that, you're still left wondering whether the structure you're looking at bears any resemblance to how the protein actually exists in its native membrane environment.
Consider two stories of MPMP’s, and their associated painful stories of working with them:
- GPR40 is a GPCR highly expressed in pancreatic beta cells, playing a crucial role in glucose-stimulated insulin secretion. This makes it a highly attractive target for type 2 diabetes, but, unfortunately, developing drugs against GPR40 has been plagued with difficulties. Including, but not limited to, difficulty of stable purification, difficulty of making it water soluble, and difficulty of using them in standard binding assays, These challenges are undoubtedly part of the reason that despite years of research, only one GPR40-targeting small-molecule drug, Fasiglifam, even reached phase 3 clinical trials. It has, unfortunately, since been discontinued due to liver toxicity concerns.
- P-glycoprotein (P-gp) is an efflux transporter, another class of MPMP, responsible for pumping foreign substances out of cells. This is a major cause of multidrug resistance in cancer, as P-gp can effectively remove chemotherapy drugs from tumor cells. Developing inhibitors of P-gp has been a long-standing goal in cancer research. Yet, this too has failed, partially due to the difficulty of working with the protein. It's extremely difficult to purify in a stable, functional form outside of its native membrane environment (which is, funnily enough, a fact unique to human P-gp! Rodent P-gps are far more stable). As a result, structural studies have been incredibly challenging, with the first high-resolution structure of human P-gp only being reported relatively recently (2018), decades after its discovery in 1976. There is, as of 2024, no approved drugs that successfully inhibit P-gp.
Of course, drug development efforts are rarely stymied by a single reason alone! It is rare that a protein simply being ‘hard to stabilize’ outright ends a program —especially because a potential solution to the above problems is to simply do whole-cell screening (which has its own challenges!) — but it certainly doesn’t help.
The tragedy is that while MPMP’s are one of the most difficult protein structures to study, they are often incredibly good targets. This shouldn’t be a surprise if you consider that their dysfunction has been implicated in a wide variety of biological pathways. Pharmaceutical companies have obviously already taken note of this and, as a result, MPMPs make up ~40% of currently known drug targets, despite them being 23% of the human proteome; a testament to their clinical relevance.
Yet, only two approved antibodies target them: Mogamulizumab, targeting CCR4 for lymphoma, and Erenumab, targeting the CGRP receptor for migraine prevention. And while there are a far more approved small molecules that target MPMP’s (20%~), antibodies can be a fair bit more efficacious for some targets, so we’d ideally like to rely on that modality as well.
All this adds up to a depressing situation: MPMPs are incredibly important, valuable drug targets, but our ability to develop protein-based drugs against them is severely hampered by our inability to work with them effectively.
What can we do about this?
Well, one more note before we move on, because I also had this question: could you simply...not deal with the MPMP at all, at least not in their entirety? Don’t MPMPs have extracellular (read: soluble) domains that can be expressed and studied in isolation? The membrane-spanning regions might be critically important for the protein's native function, but they're irrelevant if your goal is simply to bind and block (or activate) the protein from the outside. We could just use those, and happily run our binding assays and structure determination and whatever else!
And the answer is…that my assumption is wrong. MPMP’s, in fact, rarely have nicely structured extracellular regions. Single-pass membrane proteins certainly do! But the extracellular bits of multi-pass membrane proteins, unfortunately, noodles of proteins that are similarly a nightmare to work with.
Okay, now we can move on.
Computational design of MPMP proxies
Here’s an idea: could we not simply redesign our messy, non-soluble MPMP’s to simply…be soluble? The answer has, historically, been ‘yes, but it’s hard.’ In 2004, someone did it for a bacterial potassium channel protein (KcsA) In 2013, another group did it for the human mu opioid (MUR).
But it’s also kinda…bespoke. There’s a lot of custom design, a lot of thinking about interatomic potential energies of this one specific protein, and so on. Very little of the work from any one paper study on a protein seems to easily translate into another protein. This is a problem, given that there are on the order of several thousand potentially useful MPMP’s, and we’d ideally like to not spend graduate student years on creating soluble analogues of each one.
Is this possible to automate?
There is a paper from May 2023 that suggests it is! It is titled ‘Computational design of soluble functional analogues of integral membrane proteins’, which has some big names on the author list: Martin Pacesa (BindCraft), Justas Dauparas (ProteinMPNN), and Sergey Ovchinnikov (he’s Sergey). What exactly do they do?
The pipeline starts with a target structure of some membrane protein and makes an educated guess at an initial sequence based on secondary structure preferences — alanines for helices, valines for β-sheets, and glycines for loops. They then randomly mutate 10% of these positions to introduce diversity. This sequence then gets fed into AlphaFold2, which has a composite loss function that measures how well their current sequence's predicted structure matches the target. From this, we generate gradients that tell us how to modify the sequence to get closer to our desired target. These gradients update a position-specific scoring matrix (PSSM), which was then used to update the sequence again for another round of structure prediction. This is done 500 times and is also referred to as AF2seq.
At the end of process, we have a sequence and structure pair. What should we expect from this sequence?
Initial designs by AF2seq exhibited high sequence novelty compared with natural proteins and a low fraction of surface hydrophobics; however, none could be expressed in soluble form.
Well, the Alphafold process resulted in few surface hydrophobic residues, getting us slightly closer to a soluble protein, but experimentally still not what we want. At this point, the authors redesign the sequence using a version of ProteinMPNN trained with soluble proteins (solMPNN). Why not just the usual ProteinMPNN? For the same reason you might expect:
We attempted to optimize the sequences using the standard ProteinMPNN model, but the resulting sequences consistently recovered the surface hydrophobics, probably owing to the similarity of the topology to that of membrane proteins encountered during training.
And presto, we have a pipeline for creating soluble versions of any arbitrary protein! They tested this on GCPR’s, alongside a few other membrane proteins, finding that 1) a high-confidence soluble sequence for a GCPR could be found, and 2) identical (and important) structural motifs on the nonsoluble version could be found on the soluble version.
Why not just let solMPNN redesign everything and skip the Alphafold2 step? It’s a fair question and one I don’t have an answer for. One reason may be that having the ability to modify the structure slightly (via Alphafold2) to account for the inevitably structural deviations when going from nonsoluble → soluble is helpful before solMPNN redesign, but that’s just a guess.
Edit: Martin (one of the lead authors) saw this article and answered the question! Here is his comment:
The reason we use AF2seq+MPNN is because if you use only MPNN it will keep the hydrophilics in the core. Then hydrophilics outside+hydrophilics inside = collapsed protein.
Importantly though, they didn’t show that these soluble versions of membrane proteins were actually good for anything! In the end, that’s what we really care about, that the soluble, easy-to-work with version of the MPMP can actually help accelerate biological research in the dimensions we care most about. They do briefly touch on this topic as an area to explore in future research though.
Another exciting perspective is the creation of soluble analogues of membrane proteins that retain many of the native features of the original membrane proteins, such as enzymatic or transport functions, which could greatly accelerate the study of their function in more biochemically accessible soluble formats. Similarly, this would be critical to facilitate the development of novel drugs and therapies that target this challenging class of proteins, which remain one of the most important drug targets.
Prescient on their end!
Because someone did end up testing this out: Nabla Bio. This brings us to our next section…
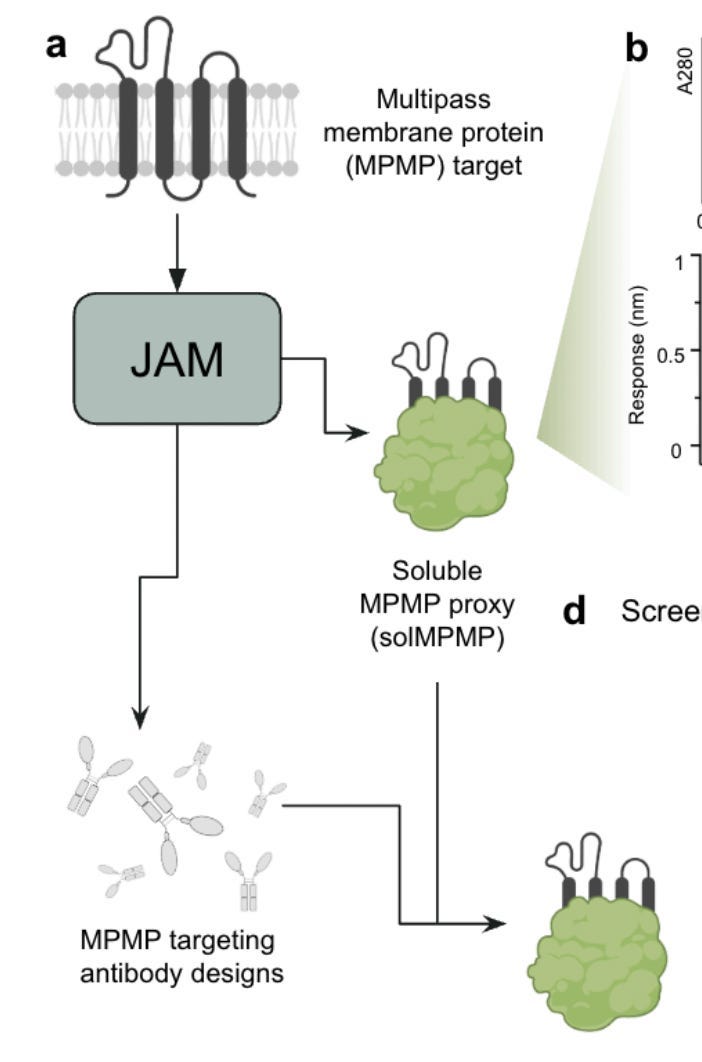
Joint Atomic Modeling
JAM, or Joint Atomic Modeling, is a technical report produced by Nabla Bio on November 11th, 2024.
What is JAM? Really, it refers to a generative model that can handle both sides of a critical problem in drug development: the membrane protein target and the antibody that needs to bind to it. When given partial information about a protein complex — whether sequence, structure, or both — JAM can "fill in" the missing pieces while respecting both hard constraints (things that must be preserved) and flexible constraints (things that provide guidance but can be modified). Pretty much no details on what JAM actually is is given in the paper, but those details aren’t super important for what we’re going to be concerned with.
The technical report includes a lot of details on the capabilities of JAM, including:
- Epitope specificity in generating VHH’s (not new, but still good to see people focusing on).
- Very, very good binding affinities in designed antibodies (sub-nanomolar range)
- Test time compute being useful for antibody engineering.
Overall, while interesting and strong in their own right, these are capabilities that aren’t particularly alien amongst many other protein foundation-y model papers these days.
But the most unique section isn’t quite about antibodies at all. As the title of this piece may have implied, it is about modeling the target. The section is titled ‘De novo design of antibody binders to two multipass membrane protein targets: Claudin-4 and a GPCR, CXCR7’. It is, as far as I can tell, the first time anyone has demonstrated the utility of machine-learned soluble proxies of MPMP’s for anything.
What do they do?
For both CLDN4 and CXCR7 (both MPMP’s), Nabla used JAM's protein design capabilities to create soluble proxy versions of them. Specifically, the transmembrane region was replaced with a stable, soluble scaffold and the extracellular structures are preserved. Which is neat! We have an all-in-one model for redesigning membrane proteins, none of this Alphafold2-ProteinMPNN pipeline stuff.
But we still haven’t gotten to connecting all of this to utility.
This is where the validation data becomes particularly interesting. For each proxy (deemed solCLDN4 for CLDN4, and solCXCR7 for CXCR7), Nabla demonstrated both structural stability (via ‘monomericity’, which refers to the percentage of protein that exists as single, individual units rather than clumping together into larger aggregates) and, crucially, functional relevance via binding to known binders:
- solCLDN4:
- 87% monomericity after one-step purification
- Maintained binding to a known anti-CLDN4 antibody
- solCXCR7:
- 85% monomericity after one-step purification
- Maintained binding to its native ligand SDF1α
!!!!!!
Functional relevance!!! Soluble versions of these multi-pass membrane proteins continue to bind to things that should bind to them!!!
But some skepticism may still be warranted: what if the anti-CLDN4 antibody and native ligand SDF1α binding is fully coincidental? If there truly was a one-to-one correspondence between Nabla’s proxies and the real protein, a screening campaign on top of the soluble protein would yield something that also binds to the native version of the protein.
And Nabla did exactly this!
For CLDN4:
In our CLDN4 de novo design campaign, screening de novo VHH designs with solCLDN4 on-yeast successfully identified three binders that exhibited EC50s of 10, 22, and 56 nM for native CLDN4 on overexpression cell lines (Fig. 6e). Among these, the best-performing binder also showed effective recognition of CLDN4 on OVCAR3 ovarian cancer cells.
For CXCR7:
In our CXCR7 campaign, screening de novo VHH designs with solCXCR7 on-yeast successfully identified a strong binder that recognized native CXCR7, achieving an EC50 of 36 nM when expressed recombinantly as a monovalent VHH in an E. coli cell-free system and tested against PathHunter CXCR7 cells.
This validation story is remarkably complete. Their soluble proxies maintained stability, bound to known ligands, and could actually be used to discover new binders that work against the native proteins. The specificity data is particularly compelling. The CLDN4 antibodies showed >100x selectivity over closely related family members (CLDN3, CLDN6, and CLDN9), despite 85% sequence identity in the extracellular regions. This suggests their proxies maintain the subtle structural features that distinguish these closely related proteins!
From start to end, here is the pipeline for this. Again, few details.
Conclusion
Typically, I’ve always ended these types of company-overview articles with a note on the potential risks of the company, but I’ll be skipping that here. Nabla is so new that it’d be difficult to give a strongly informed guess as to how they will fare. What is included in this essay is a strong validation of a thesis they are pursuing (target engineering), but not necessarily of the company at large, which, from a cursory glance of the JAM paper, is obviously concerned with a lot more than targets alone.
But what of this target engineering stuff? It’s clearly an interesting idea, feels like it should theoretically have value, and empirically works given the results of the JAM paper. How much juice is left to squeeze there?
Here’s the obvious bull case: there are only two antibodies that works with MPMP’s, partially due to how hard MPMP’s are to work with, so there’s clearly room on the table for more. If the Nabla bet really pays off, they potentially get access to first-in-class targets for a ton of different diseases or can sell access to soluble proxies to pharmaceutical partners, both of which likely have huge amounts of payoff.
But as always, there’s a bear case with these.
First, there's the question of target selection. While MPMPs represent ~40% of current drug targets, this statistic might be misleading when thinking about the addressable market for Nabla's technology. Many of these targets are potentially already being successfully pursued with small molecules. The real opportunity lies in targets where antibodies would provide meaningful advantages over small molecules, or where small molecules have failed. I don’t know the answer to how large that number is! But my guess is that it narrows the playing field a bit.
Surge, the founder of Nabla, had some thoughts on this topic of antibodies versus small molecules, which I’ve attached here:
People are generally more excited about antibodies than small molecules for a couple first principle reasons:
1. Much higher specificity. There's a lot more engineerable surface area on an antibody. This allows you to design binders with high specificity, which is critical for MPMPs, many of which look very similar to each other and thus lead to off-target toxicity/side-effects if your drug is non-specific. This is a major issue with small molecules.
2. Extended half-life: Antibodies stay in your bloodstream for weeks vs < 1 day for small molecules. This means less frequent dosing.
3. Antibodies as handles for other functions. You can use antibodies to e.g. recruit t-cells to a cancer overexpressing a GPCR or claudin, or use that antibody binding head as a CAR, or use the antibody binding head in an ADC. You can use the antibody to recruit other immune cells (important concept generally with cancer).
4. Well-trodden formulation and manufacturing path. These can be dealbreakers in DD, but for a well behaved antibody it's relatively standard. For a small molecule, it's a different process each time, and a much more frequent source of failure.
So, in the end, small molecules may be less competition than one would naively assume. There’s one more issue which is a little nuanced, and a bit out there, but it feels worth mentioning.
Returning back to Nabla’s JAM paper, when designing binders for CXCR7, they found something curious: their best binder had aggregation tendencies that might limit its developability. The authors make an observation: this aggregation propensity mirrors that of CXCR7's native ligand SDF1α. This raises a question about the fundamental nature of GPCR targeting: is there an inherent tension between effective binding and developability? The features that allow for engagement with GPCR’s may inevitably cause issues.
If this is indeed true, it may be the case that even if you can design soluble proxies of membrane receptors, in-vitro screening assays that rely on those isolated proxies will also cause issues. Here's the chain of logic:
- You create a soluble proxy of your MPMP target
- You screen for binders against this proxy
- The binders that show the strongest affinity are likely to be those that best mimic the natural binding mode
- But if that natural binding mode inherently requires "sticky" interfaces...then you're essentially selecting for problematic developability properties by design.
In the end, while Nabla may have first in-class access to targets, binders to those targets may also be awful to work with.
Is this a huge issue? I don’t think so, especially since they didn’t point out that this occurred with binders to solCLDN4. The fact that this phenomenon wasn't observed in their non-GPCR MPMP work indicates that this isn't a universal issue across all multi-pass membrane proteins.
However, for GPCRs specifically, it points to an interesting constraint on the potential of target engineering. Since whatever binders you find to that target will have downstream issues that must be amended. Of course, JAM will still be helpful! Instead of struggling with target protein stability and assay development, drug developers will be wrestling with optimizing developability properties of their hits, which instinctively feels like a faster problem to iterate upon. Overall, this point is very much splitting hairs, but maybe an interesting thing to think about.
That’s it for this piece! Excited to see what else comes out of Nabla.
0 comments
Comments sorted by top scores.