A primer on the next generation of antibodies
post by Abhishaike Mahajan (abhishaike-mahajan) · 2024-09-01T22:37:59.207Z · LW · GW · 0 commentsThis is a link post for https://www.owlposting.com/p/a-primer-to-the-next-generation-of-antibodies
Contents
Introduction What’s wrong with antibodies? Production demands Storage Efficacy What does a better antibody look like? Single-chain variable fragment (scFv) Advantages (scFv) Disadvantages (scFv) Nanobody (VHH) Advantages (VHH) Disadvantages (VHH) Antibody mimetics Advantages (mimetics) Disadvantages (mimetics) Conclusion None No comments

Introduction
If you want a primer over antibodies, I recommend reading my last post! This one will contain some jargon that the other post will explain.
It's important to remember that antibodies aren't inherently special, proteins are just strings of amino acids, and the shape of a protein is all that (mostly) matters. One can imagine a world in which we ditch full antibodies entirely and instead work on protein modalities that improves upon it; reducing their downsides and improving on what they are already good at.
The medical world focused on antibodies for an obvious reason: it clearly works well for the adaptive immune system of every single multicellular organism out there, which is a pretty strong endorsement of its clinical utility. But the pressures under which antibodies evolved are completely different to the pressures of our medical system, which is far less tolerant of extreme complexity, more interested in scalable production, and is equally interested in both the short-term + long-term quality-of-life of a patient. Moreover, our understanding of biology is rapidly advancing to the point where we can look beyond the tools that evolution has provided.
But over the next decade, where will we expand? In this post we’ll go over what is wrong with full-length antibodies and three potential alternatives to them:
- scFv’s. An older entry in the antibody engineering field, with 9 drugs released under this class of antibody, but still relatively new in terms of the antibody world.
- Nanobodies. The most exciting current development in the antibody field, with only one released drug in this category and many more potential ones.
- Antibody mimetics. Where I believe the future is heading.
One quick note before we move on. People more familiar with antibodies may wonder why I’m not discussing Fab’s, or chimeric antibodies, or bispecifics, or trispecific antibodies, or any one of the many other varieties of antibodies out there outside of the above three. This is because the above scFv’s, nanobodies, and antibody mimetics are very much in a clinical gray area; very studied from an academic perspective, but the medical impact is still badly understood. All others largely fall into the bucket of so-old-that-they-aren’t-really-next-generation or so-new-that-it’s-challenging-to-assess-how-valuable-they-will-be.
What’s wrong with antibodies?
Motivating the question here fully is important: why fix something that isn’t broken? Well, there are a few things that are broken about antibodies.
Production demands
Antibodies have an extraordinarily difficult production process. Here’s a breakdown (warning, long).
You first need to find the genetic sequence that makes each of an antibody chain (heavy and light, so 2 unique sequences). You then take these sequences and insert it into an expression vector, which is a circular piece of DNA. The expression vector is then washed over a cloned mammalian cell line, most commonly Chinese Hamster Ovary (CHO) [1] cells. The vector is able to enter the CHO’s and directly integrate into their genome. But, while these cells are often happy to produce the protein of any gene that wanders into its genome, they may still vary in ability to produce that protein. This can be for a lot of reasons. Maybe the vector ended up in a section of the genome that is rarely read from, so nearly zero antibody will be produced. Maybe some CHO’s randomly mutate, a phenomenon called clonal variation, which reduces ability to produce antibodies.
In any case, you’ll need a way to select high-performing cells. There’s isn’t really high-throughput way to do this, you literally just grow the CHO’s in small cultures and check the antibody levels in each cluster every now and then using techniques like ELISA. Let’s say you stumble across cell that produce antibody well AND is consistent, implying its output will be predictable over the long term. Now you just need to clone these high-producing cells, which can take weeks of careful preparation, and now you’re finally ready to scale up!
At this point, you can take your vast supply of high-producing CHO’s and dunk them in a bioreactor, a massive steel tank with controlled oxygen, temperature, and pH levels, and let the CHO’s produce antibodies into the growth medium it’s immersed in. But, because mammalian cells are immensely fragile, antibody growth may stall at any time. Maybe the pH of the bioreactor is off, or microbial/toxic contamination occurs, or maybe the CHO’s simply mutate, or some other unknown other reason! If there are any deviations, adjustments need to be made to the bioreactor conditions to get the cells back on track. Mammalian cells are notoriously fickle with their preferences and die easily, so this process may take awhile. If everything goes right, the surrounding growth medium of the antibodies will slowly become heavily enriched in free-floating antibodies, eventually reaching a desirable concentration. Now it’s time to harvest!
This medium, while enriched in antibodies, also contains a complex mixture of other proteins, nutrients, and CHO-produced debris that needs to be removed. This is where the purification process comes in, usually relying on a technique called ‘affinity chromatography’, which allows us to isolate the antibody via finding something that binds to it. In practice, a protein called ‘Protein A’ is used for this, which binds to the constant heavy-chain Fc region of antibodies. This is usually insufficient for meeting the FDA’s standard for antibody purity, which is 95%, and subsequent purification steps are required, such as ion exchange chromatography (IEX) or hydrophobic interaction chromatography (HIC). But let’s assume we only need the first step and move on with our liquid filled with pure antibodies.
We’re nearly done. Now, we just need to filter the purified antibody solution to remove any remaining particulate matter or aggregates. This is typically done using a series of filters with decreasing pore sizes, down to 0.2 microns, which is small enough to remove most bacteria and other small contaminants. The filtered antibody solution is then concentrated to the desired level, usually using diafiltration to increase the antibody concentration even further. We mix the resulting hyper-concentrated antibody collection in with a buffer to control pH, salts to control tonicity, and stabilizers like sugars or surfactants to prevent the antibody from degrading or aggregating during storage. The sterile antibody solution is then filled into the final containers, which are often vials or syringes.
Throughout the entire manufacturing process, from the initial cell culture to the final packaging, strict quality control measures are in place. Samples are taken at various stages and tested for purity, potency, identity, and safety. Any deviations from the specified parameters can result in the rejection of the entire batch, which means you have to start over from scratch. Moreover, each step of the process post-bioreactor has inefficiencies; chromatography and screen filtering and diafiltration all slightly reduce the yield of the final product.
Given all this, it’s no wonder antibodies are extraordinarily expensive drugs; even the generic version of the widely-used antibody drug Humira can still cost $1k~ a month at the lowest end. To compare this to typical small-molecule drugs, generic versions of Keppra, an anti-epilepsy drug, can cost less than 10 dollars per month. Antibody production is uniquely challenging and costly in a way that very little else in drug manufacturing is.
Let’s ask some questions.
Why can’t we simply synthesize antibodies, much like how we synthesize typical drugs, and avoid this whole bioreactor thing? Antibodies are among the largest and most complex molecules used as therapeutics. They are composed of four proteins chains linked together by disulfide bonds, each chain is intricately folded into specific domains, which together form the characteristic Y-shape of an antibody. And synthesizing such a large, precisely folded protein from scratch is simply beyond our current capabilities. Modern chemical protein synthesis is typically limited to peptides of less than 100 amino acids, while each antibody chain is 200-500 amino acids long. Even if we could synthesize the individual chains, getting them to assemble and fold correctly into a functional antibody would be nearly impossible. Cells, on the other hand, have evolved to do exactly this.
If mammalian cells are so fragile and finicky, why can’t we find a better cell line to produce antibodies? There is in fact a cell line that is much simpler to work with: yeast. Yeast is challenging to kill, replicates easily, grows fast, and is amenable to genetic manipulation. So why don’t we use it? There’s a really wonderful review paper that discusses all this. In short, antibodies require specific post-translational modifications, particularly glycosylation at a specific residue (residue 297 of each heavy chain), to function ideally in the human body. While yeast cells do have their own glycosylation machinery, it differs substantially from that of mammalian cells. Yeast tends to add high-mannose type glycans (as in, sugar molecules that contain a lot of mannose) to its produced proteins, which are not typically found on human proteins and can potentially make the antibody more immunogenic, which is obviously undesirable. Looking beyond yeast has similar issues, many types of bacteria lack any glycosylation system at all or struggle with the size of the antibody.
This all said, there is progress here, research is ongoing in ‘glycoengineering’ yeast to produce antibodies with human-like glycosylation, engineering aglycosylated antibody variants that work well, and even trying to add in glycosylation systems into bacteria. But mammalian cells are still very much considered the gold standard (for now).
Storage
Let’s say we have our set of purified and packaged antibodies ready to go in a few thousand vials. Now we’d like to ship these life-saving drugs to clinics around the globe. What other problem do we have to contend with?
Stability is the biggest one. Proteins in general are inherently unstable. These are long chains of amino acids that are folded into complex three-dimensional structures, and these structures are (usually) extremely difficult to maintain outside of their native environments. The forces that hold proteins together - hydrogen bonds, van der Waals interactions, hydrophobic interactions - are relatively weak. The immense size of an antibody only adds to this fragility, a larger size means more exposure to the environment, more failure links amongst the residues, a more complex structure to maintain. Small molecules, in contrast, are nothing like this. They rely on covalent bonds to stick together (which are much stronger than the forces antibodies use), don’t rely on any semblance of folded structure to function (so there isn’t any parallel to misfolding), and are several orders of magnitude smaller (and, in the chemistry world, smaller usually means more stable)
Well, okay, why can’t we just freeze it? Freezing is where most people’s mind would go to when the question of stability comes up, and it’s a good idea, cold temperatures reduce atom vibration + reduce reaction rates and thus increase protein stability. It works well for antibodies but having to store your drugs in refrigeration at 2-8°C (with cryoprotectants to prevent ice crystal formation of course!) does increase the cost of your antibody drug. Moreover, freezing does not completely solve the stability problem! Even at low temperatures, antibodies will still continue to undergo the usual protein degradation process (e.g. oxidation). The rate of these reactions is slowed down by cold, but not stopped entirely. This means that antibodies have a limited shelf life even under refrigeration, typically around <1 year. In contrast, many small molecule drugs can remain stable for much longer periods, often several years, even at room temperature.
The final issue here is aggregation. Antibodies, particularly when exposed to stresses like temperature fluctuations or agitation, can clump together to form large, inactive aggregates. This process is often irreversible, meaning that once an antibody has aggregated, it cannot be returned to its original, active form. Aggregation can occur at basically any point during the production and storage of antibodies, and it's a massive cause of loss of product and reduced shelf life. Why does it aggregate? Once again, the size of the protein can be indirectly implicated as a problem; there’s just so many fragile forces going on inside an antibody. It’s a wonder we can transport these things at all. Here’s one papers explanation of antibody aggregation:
…One bottleneck limiting mAbs therapeutics’ development is aggregation [12,13]. mAbs with 12 sub-domains, large hydrodynamic radii and surface areas, non-symmetrical hydrophobicity and charge distributions are prone to aggregation [14,15]. The immunoglobulin Greek-key β sandwich folding of mAbs is susceptible to edge-edge association [16]. Besides, complementarity determining regions (CDRs) of mAb responsible for antigen binding can also contribute to aggregation due to the frequent occurrences of hydrophobic and electrostatic residues [17,18]. Furthermore, the extensive hydrophobic patches on the surfaces of mAbs, especially on Fc could mediate aggregation [19,20]. These aggregation propensities are amplified by the natural bivalency of mAb. Importantly, the aggregation of mAb could be increased when administered by subcutaneous (SC) delivery in a high mAb concentration of >100 mg/mL [21]. At such high concentrations, mAbs are more susceptible to aggregation…
How is it possible that handling antibodies is so punishing when we’re filled with these things? It’s important to remember that these problems are much less of a concern in natural antibodies floating around in your bloodstream because the concentration is far lower than antibody therapeutics, which can be 100x more concentrated in terms of milligrams of antibodies per milliliter than in in-vivo.
Efficacy
Let’s say you’ve produced your antibodies, stored them, and have safely delivered them to the clinic that desperately needs them. Everything is fine now, right?
There’s one last, small thing. As mentioned, antibodies are reasonably large structures with a large molar mass. The large molar mass means that they are often incapable of diffusing throughout dense tissues, such as solid tumors, and their size means they cannot easily access tissues that have restricted entryways, such as the central nervous system. So, there are some conditions for which antibody therapy is simply not useful.
But overall, this is the smallest issue that antibodies face. When antibody therapy works, it works.
What does a better antibody look like?
Any alternative to antibodies must be able to tackle the challenges laid out in the prior sections. In short, it must display the following characteristics:
- Be easier to manufacture than antibodies
- Be easier to store than antibodies
- Be more efficacious than antibodies
Let’s go through all three of the major alternatives to antibodies and assess how well they tackle these items.
Single-chain variable fragment (scFv)
One approach to improving upon antibodies is cut out the Fc region and the constant section of the Fab region, since, really, the variable regions are the ones doing most of the antigen binding. Doing this would result in a ‘single-chain variable fragments’, or scFVs, which consist of only the variable regions of the heavy (VH) and light (VL), connected with a short linker peptide of a few amino acids long. This forms a structure that is 1/6 the size of a full antibody while (mostly) preserving the antigen-binding affinity of the parent antibody, as we retain all six of the CDR loops of the variable region.
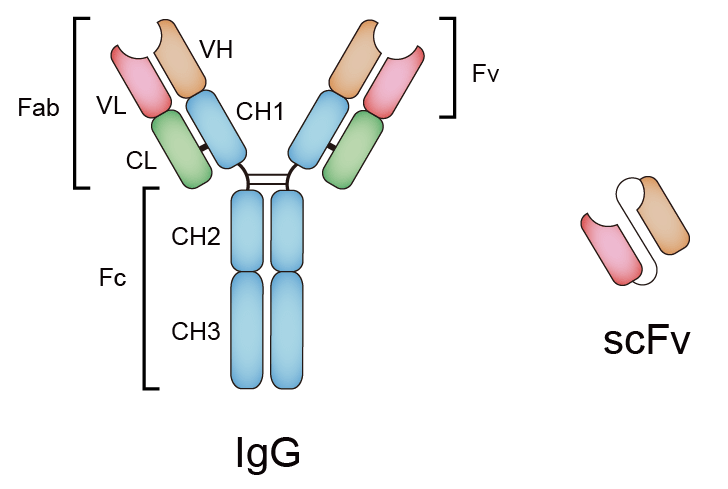
From here
Advantages (scFv)
The primary advantage of scFv’s here are claimed to be on two fronts: efficacy and ease of creation.
Because of its far smaller size (and molar mass) compared to antibodies, scFv’s can penetrate through solid tissues far more easily. In an interesting study comparing typical IgG antibodies to scFv’s (which they call sFv) ability to rapidly penetrate solid tumors, scFv’s clearly came out on top. In their words:
These studies revealed that most of the intact IgG delivered to the tumor was concentrated in the region of or immediately adjacent to vessels, while the sFv was more evenly distributed throughout the tumor mass….The sFv demonstrated maximum tumor penetration at 0.5 h postinjection, while the intact IgG reached an equivalent degree of penetration at 48 to 96 h postinjection.
Prior studies have also shown that scFv are much more rapidly cleared by the body compared to typical antibodies, potentially massively reducing the side effects of any scFv drug. Moreover, lacking the Fc region means that scFv drugs get to avoid ‘antibody-dependent cellular cytotoxicity’, potentially also massively reducing the cytotoxic effects that usual antibodies can sometimes have. This is all while retaining the usual binding capacity of typical IgG’s to its designed-against antigen.
But, I’m going to be honest, the efficacy claim-to-fame of scFv’s is a little bit suspect. Most papers trot out the same line of scFv having much better pharmokinetic profiles compared to IgG antibodies, but…I’m finding basically zero control studies on the subject. There are lots of scFv only papers studying scFv phenomena, but nobody ever pairs it up with an IgG antibody to study the exact differences. The above quote is the only one I could find and even that isn’t necessarily about efficacy, just a proxy of it! It’s genuinely strange, I have to imagine that these comparison studies exist for clinical trial purposes, but basically all scFv papers I’m finding are scFv only, like this. Please let me know if I’m missing something significant here!
Here’s something potentially interesting though: there is a singular released scFv drug, brolucizumab. Its primary competitor is aflibercept, both are intended for treatment for age-related macular degeneration. Aflibercept is technically a fusion protein, composed of two binding regions of a proteins fused with…drum roll...the Fc portion of the human IgG1 immunoglobulin. So, not exactly an antibody, but not the worst comparison in the world to see how well an scFv compares. And here are the phase 3 results for brolucizumab versus aflibercept. While it does prove ‘non-inferiority’ for brolucizumab, it’d be tough to say that it goes far part aflibercept results.
At the absolute most, I currently think that scFv’s are primarily useful in getting into parts of the body that larger (full antibodies) cannot, like the central nervous system.
The much, much larger advantage with scFv’s is in production; they do not need mammalian cells, they can be produced by bacterial colonies.
This is an absolute gamechanger. scFv’s don’t require complex glycosylation due to lacking an Fc region, are small enough such that small microbes can pump it out of themselves, and have a simple enough fold that production levels can remain high even with the simpler cellular machinery in non-mammalian cells. Because of this, most people use E. coli for scFv production. Being able to use something like E. coli to produce one’s drugs is an immense boon. E. coli is extremely cheap to grow, is not finicky to its environment, has the same level of genetic malleability at CHO’s (and maybe even more!), and is capable at growing at extreme scales (in 1000+ liter tanks) easily. One still must go through the same process of transfecting E. coli cells, selecting high-producers, cloning them, incubating them in a bioreactor, and purifying bioreactor medium to grab out the scFv’s. The primary changes are that your producers are much easier to keep alive and much faster to replicate, which dramatically speeds up the main bottlenecks in the drug creation process. Of course, there are some mild downsides; E. coli is still, strictly speaking, a worse medium for creating any large-ish complex protein than mammalian cells due to both its size and genetic simplicity. As such, e. coli may struggle with correctly folding producing the heavy and light chains, necessitating an expensive chemical process to encourage refolding. Overall though, it seems like the upsides heavily outweigh the downsides.
Disadvantages (scFv)
On the side of storage, scFv’s don’t do well. scFv’s are much more prone to denaturation from thermal stress. This implies that aggregation is a bigger deal here as well. Why is this the case? It’s…hard to tell, most papers that discuss the stability downsides of scFv’s go into relatively little detail, but it has something to do with the constant regions of the antibody being extremely important for chemical stability of the whole antibody. Without it, things go a bit downhill. From a review paper:
Further elimination of CH1-CL pair in Fab, resulting in fragment variable (Fv), significantly discounts thermodynamic stability (Webber et al., 1995; Jager and Pluckthun, 1999b). This is presumably due to the unnatural exposure of the lower VL and VH regions, flanking CH1 and CL, where hydrophobic interaction used to contribute to the stability as a whole as well as the absence of the contribution of CH1, which controls the assembly of heavy and light chains of the whole IgG molecule (Feige et al., 2009),
There’s also a few efficacy issues as well, but it’s less clear how big of a problem they are. One of them is the downside of a scFv advantage we mentioned earlier: fast clearance from the blood stream. While quicker clearance from bloodstream due to its size can be a good thing in terms of side effects, it also means that scFv therapeutics potentially lack enough time to have any therapeutic effect. But we also saw earlier that scFv can, at least in the case of cancer, still exert a therapeutic effect despite the fast clearance. Again, I’m finding relatively little information on how much fast clearance really changes therapeutic effects.
In any case, it doesn’t matter too much. Both of these issues largely have fixes for them if needed, such as modifying the peptide linker connecting the heavy chain to the light chain to increase thermostability (they modify the peptide linker to be more hydrophilic, maybe allowing the hydrophobic bottom of the variable heavy chains to add stability) and performing PEGylation on the scFv to increase half-life in the body (which is just attaching a polyethylene glycol molecule to the scFv, a common method in drug development to increase circulation time in the body). The only issue is that any method to fix the downsides of scFv will also wind increasing the cost of it!
Nanobody (VHH)
One thing we’ve noticed from antibody engineering is that the bulk of antigen binding actually stems from the heavy-chain CDR regions. Given that, we could also attempt to remove the variable light chain from the scFv, creating what’s also known as 'single-domain antibody' or VHH fragments; which are typically composed of only 110~ amino acids. There is actually a nanobody drug on the market, Caplacizumab, first approved by the FDA in 2019. To offer a comparison, the first FDA-approved antibody drug was released in 1986.
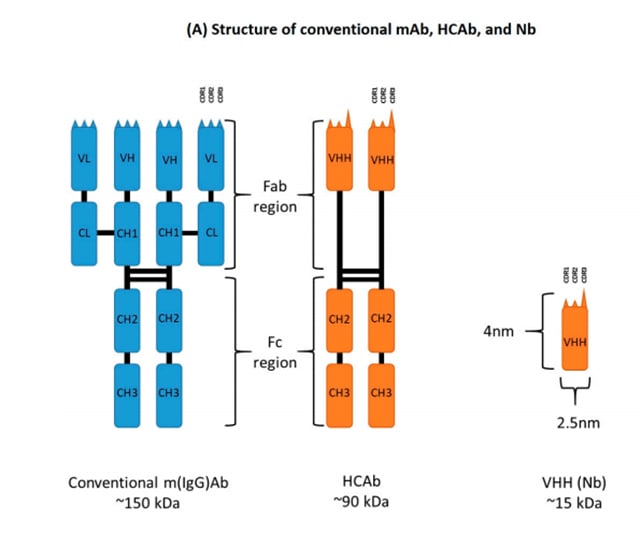
From here. 'Heavy-chain-only antibodies', or HCAb's, are found in nature in camelids and were the inspiration behind VHH's (nanobodies)
Advantages (VHH)
Basically all the same as the advantages from scFv’s, but there’s a surprising amount more we gain we get from shearing off the light-chain!
Let’s start with the least impacted: efficacy. We notice similar advantages as scFv, faster tissue diffusion, faster blood clearance, and ability to cross/touch antigens that typical large antibodies cannot, such as hidden epitopes in viruses or antigens within the blood brain barrier. But, again, it’s hard to tell the true clinical impact here, most studies assess these characteristics independent of actual clinical benefit.
There is a much larger impact on the production end. Again, all the main advantages from scFv’s carry over: there’s no need to use mammalian cells. But there’s more: not having to deal with an extra chain means e. coli becomes a degree more efficient in producing the drug, the higher stability of the protein (discussed later) means misfolding/aggregation cases are rarer, and the smaller size of the nanobody also means microbial colonies can more efficiently excrete it out.
But the most impacted by far is stability. Nab’s are extraordinarily tough, exhibiting vastly improved thermostability, pH variability resistance, reversible misfolding, and lower aggregation compared to even typical antibodies. One review article said the following:
Nbs are more resistant to chemical denaturants and protease enzymes [40] and have higher stability under harsh PH or ionic strength [41]. This higher conformational stability also stems from the presence of an extra disulfide bond, which lowers the probability of heat-induced aggregation and limits VHHs flexibility [42,43,44,45,46,47]. Because of higher stability, they show high refolding efficiency, which means raising or lowering the sample temperature does not affect Nb conformation, i.e., it de-binds and binds to the target, respectively, without any aggregation or denaturation [48]. This rigidity in structure is a favorite property in the clinic since non-native protein aggregation is a common downside of antibody treatment, raising the immune response in severe cases [49, 50].
Nanobodies are so strong that they don’t even need refrigeration! This was the subject of extreme interest during the peaks of the COVID epidemic, with one study in particular finding that their isolated nanobodies could be be freeze-dried and aerosolized with zero loss in potency. One could imagine nanobodies being used for all sorts of diseases in a much cheaper manner because of this, perhaps being given even in inhalers. Surprisingly though, looking this up yields relatively relatively little beyond more SARS-CoV-2 stuff, and basically all work into this stops from 2022 onwards. Hard to find a reason for this, potentially there is some hidden flaw in nanobodies here that I’m missing…
One quick question before we move on: how exactly does going from IgG to a single Fab chain (scFv) reduce stability, but cutting off the constant parts of that Fab chain increase it? There are a wide range of structural reasons why. Some extra bonds are created as a result of dropping the light chain, some loops are extended in a more stable way, some hydrophobic residues are better able to be packed away, etc. There’s no singular thing that’s driving the massive stability, just a bunch of small things adding up.
Disadvantages (VHH)
The only real disadvantage of nanobodies is on the efficacy front; the faster bodily clearance can be issue for some diseases. Derek Lowe has a nice essay about the single nanobody drug released, caplacizumab, where he writes…
…but realizing the potential of nanobodies was, as they say, nontrivial. They tend to have shorter half-lives than their full-sized cousins (some of which are spectacularly long-lived after dosing), and their smaller size has an inevitable trade-off in potency. In a head-to-head competition against a monoclonal, they're probably going to lose, unless you've got some specialized edge working for you.
That’s really it! One might ask then, what’s stopping people from adopting nanobodies given the extreme advantages and relatively minor downsides? One reason is that this clearance rate problem is so high (without any obvious solutions) that it simply isn’t worth it compared to most existing typical antibodies. But, as another hypothesis, it may also lie in the fact that nanobodies are simply high risk; antibodies are already an expensive therapy to develop, so the friction to shift may simply be insurmountably high.
Antibody mimetics
There is something beyond anything that even slightly resembles antibodies: antibody mimetics, which refer to any protein that can bind to antigens, but lack any structural similarity to antibodies. Antibody mimetics represent the 'fourth generation' of antibody engineering, following polyclonal antibodies, monoclonal antibodies, and antibody fragments (such as scFv’s and nanobodies).
This topic will be a little stranger than the prior two, because antibody mimetics come in all sorts of categories; there are affilins, affimers, DARPins, monobodies (still unrelated to antibodies!), nanoCLAMPs, optimers, and many more. Each of them are all built off known protein scaffolds or motifs, such as DARPins coming from a 33-residue motif called ‘Ankyrin repeats’, and undergo typical antibody engineering processes to optimize their binding to desired antigens. Moreover, there isn’t even clear agreement on what is an antibody mimetic, some studies claim some drugs as mimetics, others place them in different categories. As such, we’ll discuss them from the perspective of ‘what are the general trends amongst antibody mimetics’ and not offer too much specificity.
There is only one released antibody mimetic drug: Ecallantide, which uses a Kunitz domain as its scaffold, and was FDA-approved in 2012. One may notice that this is far before the first nanobody drug released in 2018, so how could antibody mimetics be considered the ‘next step’? This is a subjective decision on my end; it feels very much like the full scope of potential that mimetics have has not at all been sufficiently mapped out, whereas it has been a fair bit for nanobodies.
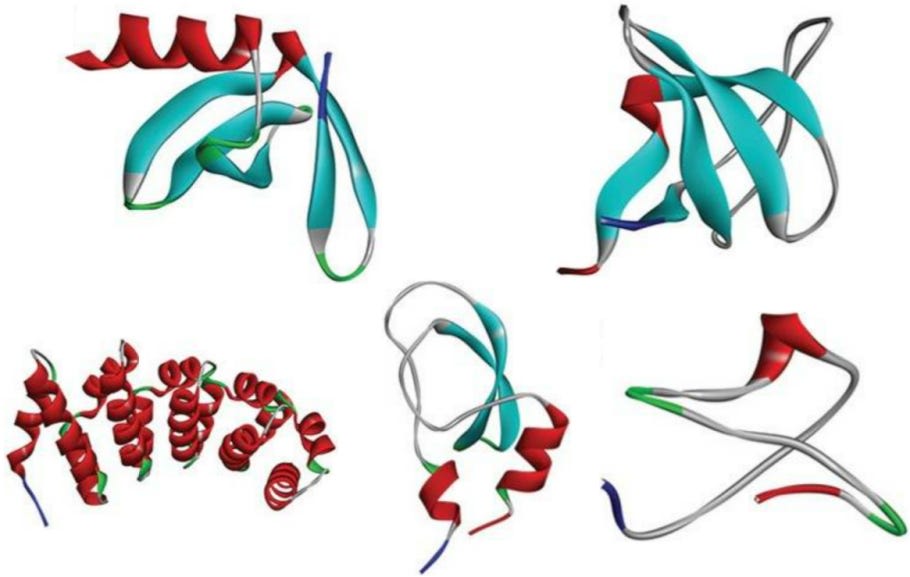
The structural diversity that mimetics can have! From here
Advantages (mimetics)
We see, generally, the same advantages as we do for scFv and nanobodies.
The storage angle is great: most antibody mimetics are based on highly stable protein scaffolds, making them more resistant to harsh conditions such as extreme pH, temperature, and presence of proteases. The efficacy angle is present as well, given that antibody mimetics can be even smaller (up to half the size) than nanobodies, they potentially allow better tissue penetration, allow access to cryptic epitopes and improve delivery to target sites, all while retaining antigen-binding efficacy. But, as previously seen, this is something that is more-so claimed than really heavily tested. Finally, the production angle is great as well; since antibody mimetics are small, single-chain, and often based on natural proteins, they can be easily produced in bacterial or yeast expression systems.
But why switch over to mimetics instead of the real deal?
Another benefit of mimetics, which scFv’s and nanobodies lack, is their flexibility (in terms of functionality, not structure!). Antibody mimetics have a staggering array of shapes and can be purpose built to do almost almost anything. Using scaffolds with a low residue count may allow you to perform chemical synthesizing of the mimetic, cutting costs by another order of magnitude. The stability of mimetics may allow you to push things even further than nanobody-level stability, potentially allowing for orally-dosed antibodies, able to brave stomach acid pH conditions. The options here are vast, and it’s really only recently that they have started to be explored.
Disadvantages (mimetics)
Again, the same clearance issues as we see in any protein that’s small.
But there’s another, more subtle one. Antibodies, even the fragments, are a well-trodden therapeutic target. Decades of research have gone into understanding the structure, function, and clinical applications of antibodies.
In contrast, antibody mimetics are a relatively new class of therapeutics, and each mimetic platform (e.g., DARPins, Affibodies, Anticalins) has its own unique characteristics and challenges. While some platforms have made significant strides, it largely pales in comparison to how much we understand antibodies. Everything from discovery, optimization, manufacturing, regulatory approval, and clinical adoption may end up being a mess, the whole therapeutic modality is still a story that has yet to go beyond the first few chapters. This may mean, despite the potential they have, mimetics will be more costly, harder to produce, and harder to understand for a long time to come.
Conclusion
Unlike my other posts, the ‘story’ here is hard to unravel, lots of things are still opaque. If scFv’s are so hard to keep stable, how have they racked up 9 drugs based on them? Nanobodies seem incredible on the surface, but why have they stalled in clinical development for decades; were the clearance problems and risk issues really that big of a deal? If mimetics are really the next generation of therapeutic, why does it feel like no one is strongly focusing on them? This feels like the nature of discussing any topic touching on clinical science; negative results are hidden or never written about, the complexity of drug development become even more apparent, and unknown unknowns ramp up.
Here’s one version of the story: there is no superior modality here. For all the disadvantages IgG antibodies have, they have by far positively affected millions of more lives than any other modality on this list. It may very well be the case that Fc mediation is important for many therapeutics, and if you lack this immune-system crosstalk, the utility of most antibodies disappear. And if we solve the biggest bottleneck of antibodies, its dependence on mammalian cells, maybe this so called ‘next generation of antibodies’ will never truly come to pass. They may be used for certain, hyper-specific diseases, but there will never be a Humira-esque blockbuster of a drug amongst antibody alternatives.
But there’s another story we could tell, one that is on shakier ground, but a lot more interesting to think about. Potentially, antibodies, as a functional category of therapeutic, may very well be on their way out. scFv’s and nanobodies may take us a bit further, but even that may disappear. Sticking with an evolutionarily-derived protein only gets you so far, and the space of possible biology is staggeringly vast — it feels extremely unlikely that antibody-like structures is the best we can do. The future very much looks like antibodies mimetics; custom-built antigen-binding protein able to be precisely tuned for their exact task, no attachment to shapes or binding sites, only focusing on efficacy, stability, and ease of production. But over what time horizon will this happen? Given the historical precedent of nanobodies, a drug modality with also a fair bit of promise taking nearly 30 years to reach the clinic, it’s hard to tell. It may be the case that the medical community moves closer to antibody fragments over the years, such as scFv’s and nanobodies, but keep a wary eye on mimetics; waiting until there’s enough evidence to finally push on it.
But this story may end up playing out differently than historical precedent suggests, because the era of nanobodies did not have one thing we have today: ML-based de-novo protein design that is getting better month-by-month. The immense de-risking that this provides may rapidly speed up our evolution from typical antibodies and make drug companies embrace modality sooner than later. In such a world, drugs that work as well as usual antibody drugs may become far cheaper, more effective, and easier to transport to those who need it most. Antibodies and even antibody-fragments become part of a bygone era of drug development, only used for very specific diseases and conditions.
Or maybe not! Feels like forecasting anything accurately in the drug development space is an exercise in futility, all that we can say for certain is that the future is interesting.
- ^
Why CHO’s? It goes beyond this post, but it’s a lot of little things. They have efficient methods for secreting out large proteins, attach the correct (read: works well in humans) sugars to produced proteins, and are amenable to genetic manipulation.
0 comments
Comments sorted by top scores.