A primer on ML in antibody engineering
post by Abhishaike Mahajan (abhishaike-mahajan) · 2024-09-23T17:03:07.628Z · LW · GW · 0 commentsThis is a link post for https://www.owlposting.com/p/a-primer-on-ai-in-antibody-engineering
Contents
Introduction Antibody background What is an antibody? Antibody structure Antibody-antigen binding Why would we want to design antibodies? Antibody Engineering Traditional antibody engineering Rational design Directed evolution Antibody engineering as an ML problem Datasets LLM-guided antibody mutations De-novo CDR design conditioned on an antigen De-novo nanobody design conditioned on an antigen Conclusion None No comments

Introduction
I increasingly see that a fair bit of the progress in ML-based protein design methods is occurring in antibody engineering, which is the process of creating antibodies tailor-made to bind to specific things in the body. I've noticed this phenomenon for the past year, and never quite understood what was going on in the space.
I've been recently taking the time to better understand this whole field a bit more and made a post about it to cement what I've learned. To note, this is not a machine-learning post in a traditional sense. I won't be going into the exact details of how antibody ML methods exactly work, but rather setting up the contextual knowledge needed to understand these methods at all + going through a few antibody ML papers.
Antibody background
What is an antibody?
I'm posting this picture right now so you have a place to refer back to it later. Ignore it for now, but it'll probably be useful down the road.
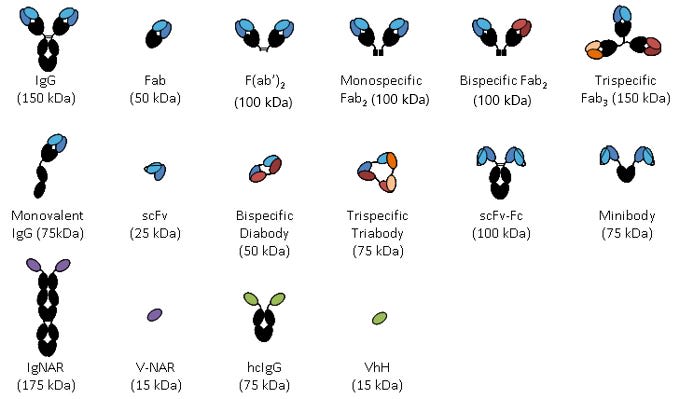
From here
The immune system is one of those things that is, unfortunately, impossible to understand unless you know all the pieces involved in it. This becomes increasingly clear whenever you look into antibodies; while a surface-level understanding is easy enough to derive from a sentence, fully grasping whats going on requires a bit more work + circling around.
Let's start with B cells.
B cells belong to the white blood cell family. Each B-cell is meant to be unique in one capacity: what they bind to. The thing they bind to is commonly referred to as an 'antigen'. Antigens can be extremely diverse, it could be the venom of an insect to fragments of the influenza virus to proteins from shellfish. Let's assume for now that an antigen is bad. This practically isn't the case, 'antigens' are a subjective category, and the human body contains trillions of 'self-antigens'. But, for now, an antigen is a bad thing that shouldn't exist inside you.
How are they binding to these antigens?
Well, most B-cells are actually identical to one another except for the part of it that binds to their unique antigen. The singular part of them that is unique and responsible for this binding is called the B-cell-receptor, or BCR. This functionality is meant to be available from the very moment a B-cell is born, it lives to bind to something, even if it doesn't know what it is. It may live its entire life never binding to the thing it was born to bind to. This will actually be the case for most B-cells in your body; there are such a staggering number of them that the antigen it was created for may not even exist in the natural world. How does the body create such a high diversity of these cells? It goes beyond the scope of this post, but look into V(D)J Recombination if curious.
But let's say the BCR does bind to something in your bloodstream. We'll brush over some steps here, but if all goes right, upon the BCR successfully binding to its own unique antigen, the B-cell will enter an 'activated' state. An activated B-cell will differentiate into 1 of 2 possible new cell states: a memory B-cell and a plasma cell. For now, we'll ignore the former case, it is largely irrelevant for the discussion of antibodies. If an activated B-cell goes the latter path, or becomes a plasma cell, it becomes a large-scale production factory of BCR's, pumping them out by the billions into your bloodstream. But, at this point, the secreted form of a BCR is no longer a BCR, but an antibody. Antibodies are simply the secreted form of a BCR, composed of only the part of a BCR that can bind to an antigen. Past producing antibodies, activated B-cells also start replicating at a massively high rate, helping ramp up even more production of antibodies.
But let's return back to the antibody-producing plasma cell, why is it producing so many antibodies that bind to the antigen that activated it? Well, we'd like to interfere with the activity of the antigen in some way. Antibodies can do just that, regardless of what exactly the antigen is. If the antigen is a bacteria or virus, antibodies being bound to the surface of it can prevent it from successfully entering a cell. If the antigen is some toxin, antibodies can prevent it from interacting from the tissue around it. Regardless of the antigen type, antibodies also serve to alert cells other than the activated plasma cell about what the antigen is and also what binds to it. We won't discuss that aspect much, but look into the complement system if interested!
So that's the life-cycle of an antibody. Now, knowing the biological context in which it exists in, we can ask 'what does an antibody look like and how does it bind to things'?
Antibody structure
For understanding antibody structure, having a visualization is basically necessary. We can view BCR's as looking largely equivalent to an antibody, sans a few extra proteins at the bottom of the antibody that allow it to transduce information about its current state to the rest of the B-cell. As a matter of terminology, you may see 'immunoglobulins', or Ig/IgG, used to refer to antibodies or BCR, it really all means the same thing. You may also see 'monoclonal antibodies', which just refer to normal antibodies that have been created from a single cloned cell, so you can have many (often billions) of the same one.
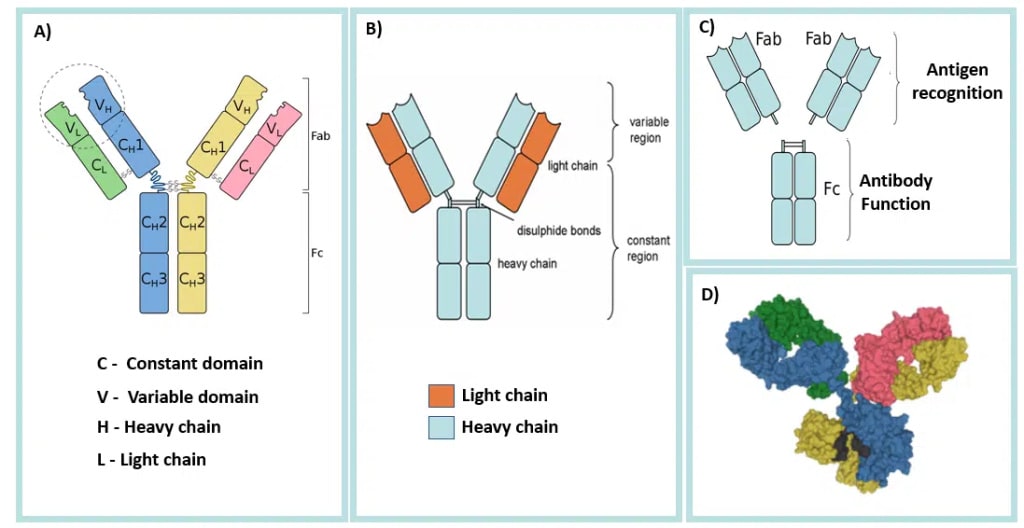
https://teachmephysiology.com/immune-system/adaptive-immune-system/antibodies/
Antibodies always come in a symmetric 'Y' shape. The top two segmented ('\' and '/') are identical to one another and really the only thing we'll be talking about. They define exactly what antigen the antibody binds to, and, consequently, this region is known as the 'Fab', or fragment antigen-binding, region. We can, for all intents and purposes, ignore whatever is going on in the 'trunk' of the Y shape, or the 'Fc' region, which stands for fragment-crystallizable region. It is extremely important, but not for antibody engineering as it stays basically the same always, it's mainly to allow the antibody to interact with other parts of the immune system.
On the tips of each of the '\' and '/' segments are 'variable' regions. This is what is usually modified to cause a change in antigen binding. To note, not all of this region is variable, only specific parts of it are (which we'll discuss later). At the base of the '\' and '/' are 'constant' regions. One word you'll often see in antibody literature is 'isotype'. The constant regions encode the isotype of the antibody, which change how it interacts with other cells (including the rest of the immune system). There are 5 known isotypes: IgG, IgM, IgA, IgD, and IgE. As the variable regions alone define what antigen the antibody is capable of interacting with, we'll largely ignore isotype entirely in this post.
The outside of each segment is composed of a 'light chain', and the inside is a 'heavy chain'. Light chains and heavy chains are disambiguated by the number of amino-acids in each one; light chains have 220~ amino acids, heavy chains have 450~ amino acids. You'll notice that each one heavy and light chain have variable regions, meaning that altering either one will change which antigen the antibody can bind to.
Finally, antibodies are usually composed of four distinct proteins, strings of amino acids. You may look at the above image and think 'arent there more? maybe 6? or 12?'. There are only four, the heavy-chain on each side of the 'Y' continues downwards. As in, the Fc region is comprised of the remaining parts of the two heavy chains that extend down from the Fab regions and join together.
And let's preempt some questions about the whole process:
Why is it Y-shaped? We don't fully know, but we have some guesses. For one, having multiple binding regions on a single antibody increases binding strength to any given antigen, or potentially allows it to bind to multiple of the same antigen. Two, the separation of the Fab regions and the Fc region is important; they are involved in binding to two completely entities (antigens versus immune system cells). Finally, the flexibility of the antibody head is important in order to allow it to bind to antigens at a variety of angles. A 'Y' shape may simply be what evolution arrived at as the optimal shape to meet all these objectives.
Do all creatures have similar-looking antibodies? Yes, for the most part. Of course, there are always exceptions in biology: there are antibodies that exist in nature that deviate from the norm. There are so-called 'heavy-chain antibodies', or HCAbs, that are found in camelids and sharks, which have the usual Y-shaped structure but no light-chains.
Can we modify antibody structure even further? Yes! We've created 'bispecific antibodies', which have two different Fab regions, allowing binding to two completely different antigens. We could also strip out the Fv region and Y shape entirely to create 'scFv' antibodies, which have only a single variable heavy chain + light chain. We've also made antibodies that have only a single heavy-chain variable region, referred to as single-domain antibodies, or 'nanobody', or VhH. However, the bulk of antibody re-engineering with AI does not bother with large structural changes of the antibody, only redesign of the variable region, so we won't discuss this aspect much...outside of one section at the end.
I'm leaving out a lot. As Derek Lowe wrote in his nanobody post (which I highly recommend!), 'in any discussion of immunology that runs to less than about 500 pages in 6-point type you'll be leaving out a lot.', but we've gone through the basics.
Antibody-antigen binding
Let's focus in our attention to the regions of the antibody that are actually binding to the antigen. As discussed before, these are part of the Fab region, and specific segments of this region are called the 'complementarity-determining region', or CDR. It may also be referred to as 'hypervariable' regions, but this seems to be less common terminology. It may be useful now to abandon the more simplistic antibody structures shown so far and now focus in on the crystallized structure of the antibody. The set of CDR's on an antibody are largely all that matters for binding to an antigen.
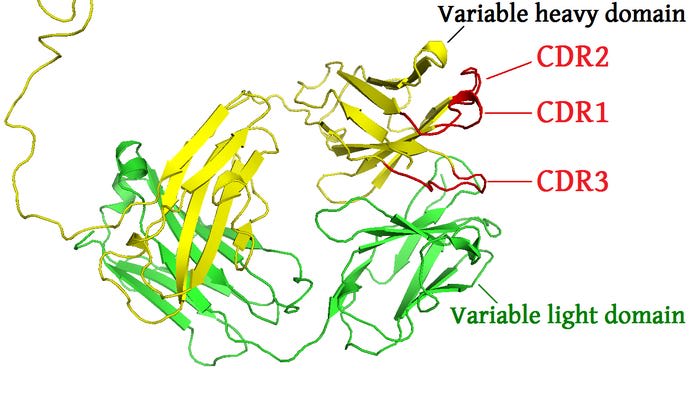
From here.
{kind=link}
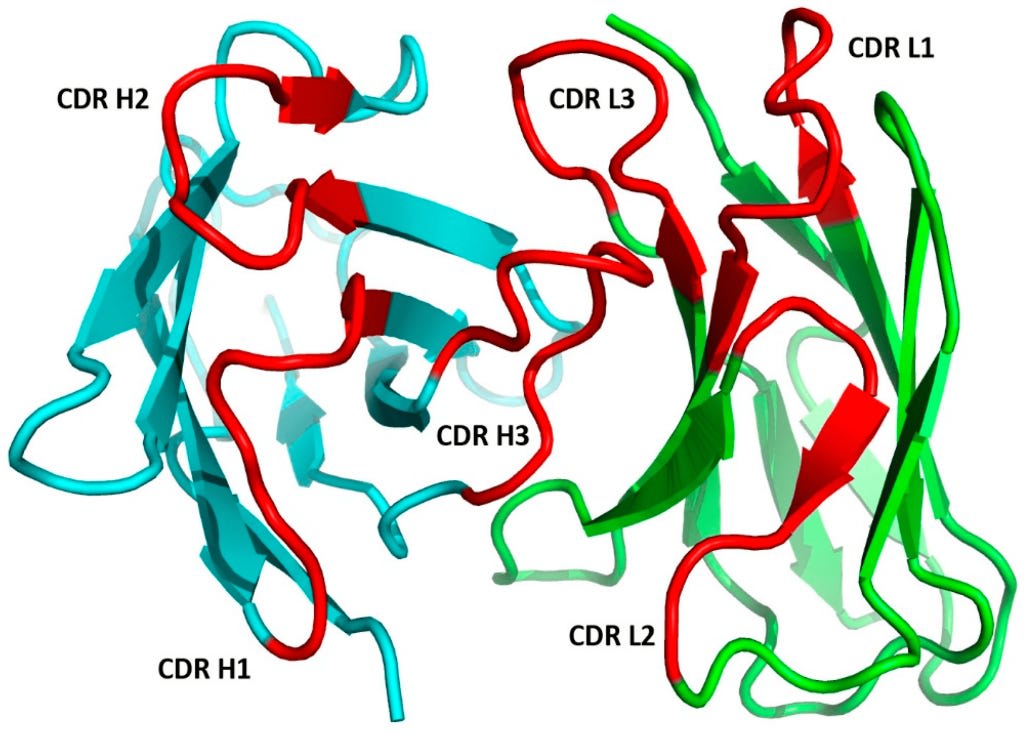
The above pictures shows the two Fab regions of an antibody. The CDR's of the heavy chain are shown in red, denoted as CDR1, CD2, and CDR3. The light chain will have three CDR's too. These are typically denoted with an H or L. So, each Fab segment will have 6 unique CDR's in total: HCDR1, HCDR2, HCDR3, LCDR1, LCDR2, LCDR3. You may also see slightly different notation depending on the text, I have also seen CDR-H1 or H-CDR1. And this will repeat for the other Fab segment, so each antibody will have 12 total CDR's. The sum combination of all of these regions make up the paratope of the antibody.
Finally, how do we refer to the non-CDR's of the variable region? As in, the region that's connected to the CDR's, but not a part of it, That's the framework region, often referred to as the 'FWR' region. These display some variability, but massively less than the CDR regions, and aren't often modified in antibody engineering problems.
Here's a question to test our understanding: how many possible paratopes are there? Given that each CDR is between 8-15 amino acids, with 6 CDR's in total, and 20 amino acids in total in the human body, that means we range from ((20^8)*6) to ((20^15)*6) possible paratopes, or 10^11 to 10^20. On a more practical level, the theorized upper limit of all paratopes is more-so 10^13, there are several practical limitations to antibodies that prevent us from reaching the true upper limit, but we won't get into that.
Here's another picture, zoomed in on a single paratope.
A quick note about antigens: while antigens can be basically anything, there is still some terminology for them. The epitope defines the region of an antigen that an antibody is capable of binding to. As one might expect, defining the space of all possible epitopes is complicated, as its dependent on the antibody used for the given antigen, the environment in which the antibody and antigen are in, and even the flexibility of the antigen (see the below picture).
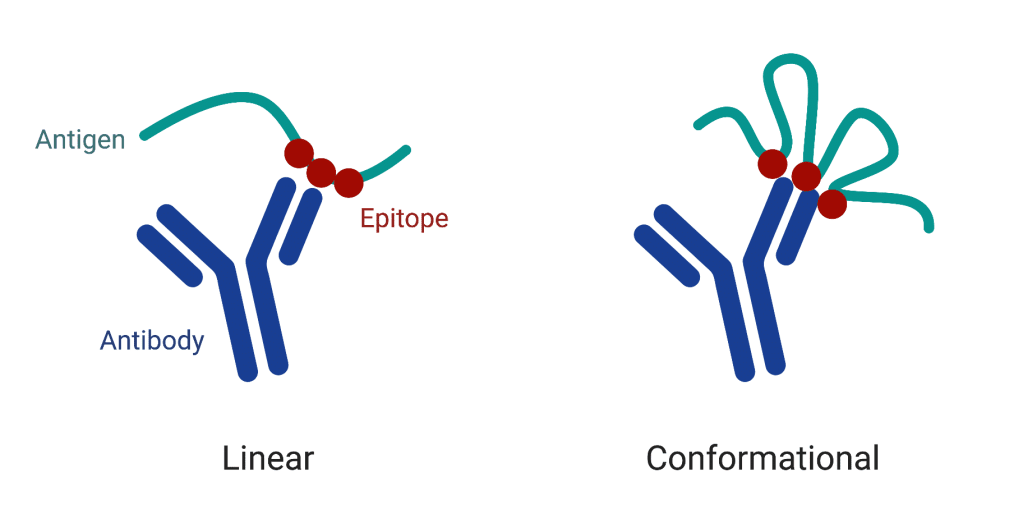
Picture taken from here. Some more reading: Conformational epitope
One immediate question we may have about this whole process is 'what do we mean by binding?'. Without getting too into the weeds, binding between antibodies and antigens are typically measured via the equilibrium dissociation constant, often referred to just as Kd value. This represents the concentration of antibody at which half of the antigen binding sites are occupied at equilibrium. A lower Kd value indicates a higher affinity, meaning that the antibody binds more tightly to its antigen. Suppose we have an antibody that binds to its target antigen with a Kd value of 1 nM (nanomolar). This means that at equilibrium, half of the antibody's binding sites will be occupied by the antigen when the antibody concentration is 1 nM. If the Kd value was instead 5 nM, that means it would take 5x the amount of antibody for half of its binding sites to be occupied. Antibodies with Kd values in the nanomolar range (e.g., 1-10 nM) are generally considered to have high affinity, while those with Kd values in the micromolar range (e.g., 1-10 µM) have lower affinity.
And this is about all the background knowledge needed. We have paratopes of an antibody, composed of 6 variable CDR's, 3 heavy and 3 light, regions that we can modify. Each CDR has between 8-15 amino acids. And all of this is meant to bind to the epitope of some antigen, the strength of which can be measured via the Kd value.
Before we further discussing the engineering of the antibody, let's quickly motivate the problem.
Why would we want to design antibodies?
Being able to custom-create antibodies to target specific antigens allows clinicians, in some sense, to take the immune system's job into their own hands. Rather than relying on the body's natural immune response, which may be inadequate or even nonexistent in certain diseases, engineered antibodies provide a way to directly intervene and guide the immune system to fight the desired target. Engineered antibodies function by the same principles as the body's native antibodies, binding selectively to their target antigen and triggering various immune responses such as neutralization, opsonization, or complement system activation.
What are some examples of engineered antibodies being uniquely valuable in a clinical context? The most obvious one is in the case of infectious diseases.
The general public became more aware of the utility of monoclonal antibodies during the peak of the SARS-CoV-2 pandemic, especially after Trump's infamous usage of the Regeneron antibody cocktail. This particular cocktail was a combination of two designed antibodies, casirivimab and imdevimab. Both of these were designed to bind directly to SARS-CoV-2, each one targeting a slightly different region of it (combinations of antibodies that bind to the same antigen + different epitopes are known to prevent mutational escape).
But why would you need these tailor-made, engineered antibodies instead of relying on your immune system to go through the antigen -> B-cell recognition -> plasma cell producing antibodies?
Here are some reasons.
- Time: Developing a natural antibody response takes time, as the immune system needs to recognize the pathogen, activate B cells, and produce specific antibodies. In severe cases, this delay can lead to serious complications or even death. Engineered antibodies, on the other hand, can be administered immediately, providing rapid protection against the pathogen.
- Affinity: The natural immune response may not always produce antibodies with high affinity, or binding strength, or Kd, for the pathogen. While I have talked at length about how your body has BCR's (and antibodies) for almost every known antigen, it isn't assured that these natural antibodies can tightly bind to an antigen, only that it can bind at all! Engineered antibodies, like casirivimab and imdevimab, are designed to have extremely high binding affinities with the antigen, ensuring that the antibody can do its job that much more effectively.
- Vulnerability: Some patients, such as the elderly or immunocompromised individuals, may not be able to mount an effective adaptive immune response at all. Engineered antibodies can, in these cases, serve to largely replace the role that their adaptive immune system would otherwise take on.
Infectious diseases are one of the more obvious places for antibody therapies to be applied. But common antibody therapies also extend into two more categories: cancer immunotherapies and autoimmune conditions.
In the former case of cancer, antibody development closely matches the infectious disease framework, only instead of viruses, the target can be certain cell receptors that are highly expressed by certain types of cancer. An example case here is Rituximab, which targets the CD20 antigen, which is highly expressed on the surface of B-cell lymphomas. Since the adaptive immune system often fails entirely to recognize the traits of cancerous cells, engineered antibodies are a vital line of therapy; though, of course, it fails entirely for cancers that mutate too fast for there to be a consistent display of recognizable cell receptors. An especially interesting direction here are 'antibody-drug-conjugates', which are antibodies engineered to bind to cancer-cell-specific receptors, but are also chemically linked to chemotherapy compounds; allowing us to deliver chemotherapy to only cancerous cells while sparing healthy ones.
The latter case of antibodies for autoimmune conditions is a bit more interesting; instead of defending from a threat, like an internal mutation or virus, the focus is on dampening down the body's own immune system in a hyper-specific way. An example here is the engineered antibody adalimumab (Humira), created to alleviate symptoms from rheumatoid arthritis (RA). The logic here is that RA drives the upregulation of tumor necrosis factor-alpha (TNF-α), pro-inflammatory cytokine that causes joint inflammation, and adalimumab is engineered to bind to TNF-α. By binding to the cytokine and preventing its action on other cells, the antibody therapy massively reduces the primary symptoms of RA. Without engineered antibodies here, a sufferer of RA only has one other instant fix: broad immune system suppressants, which are obviously undesirable.
I've shown the bright sides of antibody therapies, but given that infectious diseases, cancer, and autoimmune conditions are still a problem in today's world, it is obviously not a panacea. While there's a bevy of problems we could discuss here, there is one that is especially salient to antibody engineering: polyreactivity. Natural antibodies that exist in the human body go through an immensely complex process called central tolerance, which weeds out any would-be antibodies that react to antigens that are on the surface of our cells, secreted proteins, and so on. The reason for this is simple: you don't want an immune response to be mounted against parts of you (otherwise known as autoimmunity!). When we engineer antibodies, which have obviously not gone through this central tolerance process, we may find that they not only bind to the antigen we want, but a great deal of other, very important proteins in the human body! In such a case, we'd label the engineered antibody to be polyreactive and (hopefully) shelve it, as giving it to a patient could occur in an almost immediate (potentially fatal) autoimmune reaction.
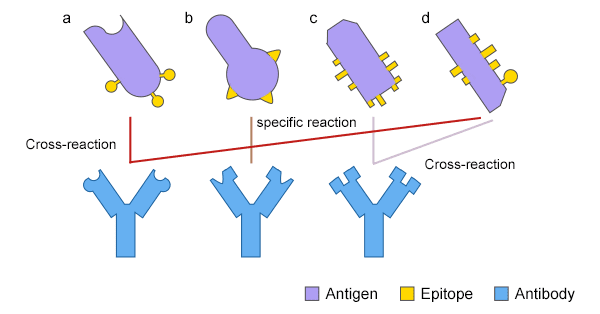
From here
Antibody Engineering
Now that we understand what antibodies are, the clinical context behind why engineered antibodies is important, and what does engineering an antibody actually involve (modifying the paratopes of an antibody to better bind to the epitopes of an antigen), we're ready to start discussing how design actually works in practice. We'll first cover how design of an antibody is traditionally done, the drawbacks of it, and then go over how ML is being applied here.
Traditional antibody engineering
Traditional antibody engineering has relied on a combination of 'rational design' approaches and 'directed evolution'.
Rational design
In rational design, researchers use their knowledge of antibody structure and desired epitopes binding to make targeted changes to the paratope. The approaches here are extremely diverse. One particularly common technique relies on crystallized structures of protein-peptides to generate peptides to graft onto CDR's. The core idea here is that if a segment of protein (say, a stretch of 7-amino-acids) are often closely found near the desired epitope of an antigen (using databases of crystallized proteins), one could simply put those same 7 amino acids in a CDR loop of our antibody to get the same binding effect! In this paper specifically, they find that multiple stretches of interactions could also be tied together, allowing you to arrive to a 7-amino-acid insertion from observing occurrence of a 3-amino-acid x epitope and 4-amino-acid x epitope. Surprisingly, this method of 'plucking out interactions' often works; distilling protein-protein interactions down to a few specific amino acid groups is definitely more an art than a science, but experienced protein designers can recognize the situations in which methods like this work. There are plenty of other methods used in the rational design space, but they all fall in these lines of 'recognize what evolution has already produced and copy it'.
Of course, this is often a painstaking process that requires years of experience to effectively do, is still often quite error prone, and is massively low-throughput.
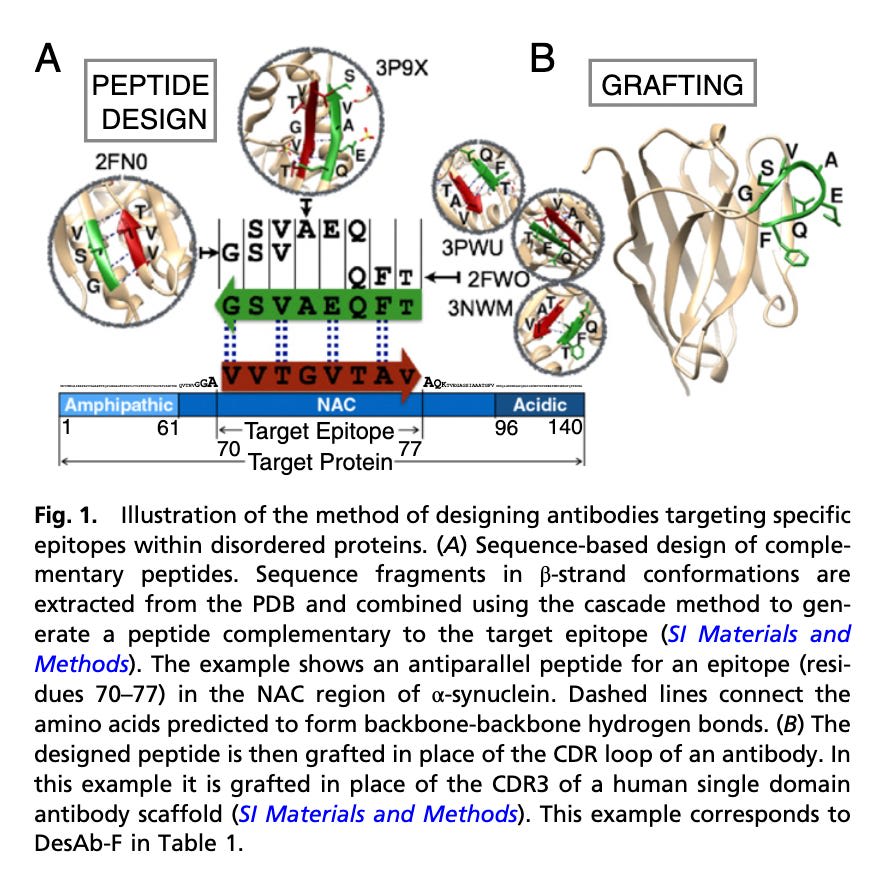
From here, picture demonstrates using crystallized structures in the antibody design process
Rational design for antibodies is often also coupled alongside molecular dynamics (MD) simulations. These simulations allow a researcher to understand how their designed antibody interacts with an epitope, how it reacts to temperature/pH changes, and how structurally stable it is, all via simulating the underlying physics of the antibody as it interacts with the world around it. Here is an example paper, where they analyze the results of MD simulations of the Fab region of an antibody designed to bind to the human protein A33, finding that the antibody is unstable (according to the simulations) at lower pH values.
However, it's unclear how accurate these simulations actually are and how much value they provide in the antibody development process. For practical purposes, it seems like simply confirm the results of wet-lab experiments and rarely provide unique information by themselves. As with rational design in general, MD is also extremely slow, with nanoseconds of protein fragments often taking hours to days of compute time, and setting up the 'correct' parameters for a simulation is very much an art honed by its practitioners over years.
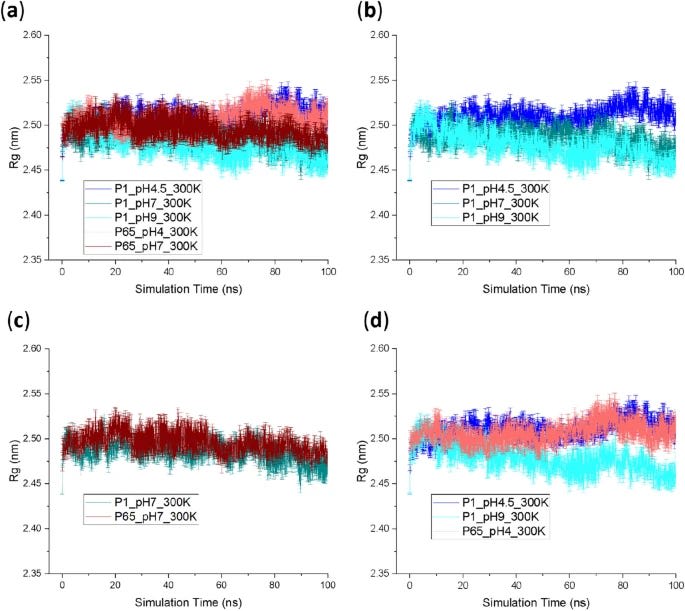
From here. Antibodies were simulated via a variety of starting conditions (pH and different starting crystal states) and their flexibility (Rg) were recorded over 100 nanoseconds.
Directed evolution
The directed evolution (DE) case is a fairly large step up in efficiency. Here, researchers start with a 'parent' antibody that already binds to the target antigen, but perhaps not with the desired affinity or specificity. They then create a library of antibody variants by randomly mutating the gene that encodes the antibody, focusing particularly on the regions that make up the paratope. These variant libraries, which can contain billions of different antibody sequences, are then expressed in phage, yeast, or mammalian cells and screened for binding to the target antigen (using techniques such as surface plasma resonance or phage display). The top performers are selected, and the process is repeated, with each round of screening and selection resulting in antibodies with increasingly optimized properties.
The drawbacks here are bit more nuanced than the rational design case.
- Bias towards high-affinity binders. Directed evolution methods tend to select for antibodies with the highest affinity for the target antigen. While high affinity is often desirable, it may not always be optimal for certain applications. For example, in some cases, moderate-affinity antibodies may have better tissue penetration or faster clearance rates, which may be considered more valuable than pure binding strength.
- Lack of control over epitope specificity. Random mutagenesis and selection based on binding affinity alone may not always result in antibodies that bind to the desired epitope on the target antigen. This can be especially an issue when aiming to develop antibodies that target specific conformations or post-translational modifications of the antigen.
- Dependence on the starting antibody. The success of directed evolution largely depends on the quality of the starting antibody. If the initial antibody has poor specificity or binds to an irrelevant epitope, the resulting optimized antibodies may not have the desired properties.
- Limited sequence diversity. Although directed evolution can generate large libraries of antibody variants, the sequence diversity is still limited by the starting antibody sequence and the mutation methods employed. This may restrict the exploration of novel antibody sequences that could potentially have better properties.
- Time and resource intensive. Directed evolution requires multiple rounds of library generation, screening, and selection, which can be time-consuming and resource-intensive. Each round may take several weeks to complete, and the entire process may require several months to a year to develop an optimized antibody.
Some may consider these to be minor problems and, in many ways, they are. At the level of billions of created antibodies, over multiple optimization rounds, at least one often meets the minimum clinical criteria. But the cost that goes into directed evolution can still be significant, especially as the number of rounds expand, and the turnaround time can be problematic for pandemics that require immediate fixes.
Is it possible for ML to handle the antibody design problem entirely and allow us to design antibodies in under an hour, with zero experimental follow-up, at any desired affinity value, with unlimited diversity, and at specific epitopes? Well...not yet, but we're getting there.
Antibody engineering as an ML problem
There are several ways that people have proposed applying ML to the antibody engineering problem. I'll first go through an explanation of the major datasets used in the field (and what they contain + value they provide to practitioners). Then we'll go through four well-known models in the antibody x ML field. Each section is meant to deepen our understanding of the problem, not to fully explain the underpinnings of each method! This field moves fast, and this blog post will undoubtedly be out of date within a few months, but the intuition we'll build by going through these papers will continue to be useful.
Datasets
There are only two main datasets here. We'll refer to them by their acronym in the following sections.
OAS, or Observed Antibody Space, is a comprehensive collection of antibody data derived from next-generation sequencing experiments. To note, sequencing is limited to the Fab region, primarily on the variable heavy-chain (VHC) and variable light-chain (VLC) regions. For a significant fraction of the dataset, some constant information is captured, allowing isotypes to be assigned as well. The primary value of this data comes from the sequences of the variable regions, which is helpful in characterizing the exact space of all 'possible' antibody sequences, especially in the CDR regions, allowing researchers to assess diversity of generated antibodies compared to natural ones. The dataset also contains data from nontraditional antibodies, such as nanobodies, but that's a minority.
- This dataset is divided into two sections: unpaired sequences and paired sequences. Unpaired sequences have the VHC and VLC sequences separated and cannot be tied back to one another. This is an unfortunate consequences of the limitations of sequencing technology; capturing sequences of proteins chained to one another (as is the case in antibodies) is challenging to do at a high-throughput scale! Advancements in sequencing technology means we can sequence paired sequences as well these days, being able to have the VHC and VLC sequences from the same antibody tied to one another, but it's at a much scale. Whereas the OAS has 3B unpaired VHC/VLC sequences, there are only 120k paired VHC/VLC sequences.
- An interesting drawback note about this dataset, and many others like it, is that it's derived from 'naive' B-cell BCR's, not raw antibodies! We've discussed BCR's, but what makes for a naive B cell? We've also discussed B-cell activation, which is when B-cells come across an antigen that binds to its unique BCR and transforms to an 'activated' B-cell, which pumps out billions of antibodies + begins replicating. What we haven't mentioned is that this replication process is intentionally error-prone in regions of the CDR, about a million times more-so than usual B-cell division. This means the antibodies produced by the 'children' of an activated B-cell are extraordinarily diverse. Most importantly, these children cells often have much higher binding affinity to the antigen it's meant to bind to than its 'parent' B-cell1. Unfortunately, activated B-cells are the minority of B-cells in your body, making sampling them by sequencing methods challenging. This leads to a sampling problem, where OAS is primarily composed of so-called 'germline' B-cells that have a ceiling on the amount of expected diversity + binding affinity in BCR's, as germline B-cells typically have low affinity for any given antigen. In the era of large-scale LLM's, this type of dataset bias can lead to potential problems, here's an interesting paper about it. Antibody AI papers will often bring up this problem as well, but it's not a huge concern for the time being.
SAbDab, or Structural Antibody Database, curates experimentally determined antibody structures from the PDB. As with OAS, more exotic forms of antibodies are also included here, like nanobodies, but it's the minority. Some statistics on dataset size are below, it's quite a bit smaller than the sequence-only dataset. This dataset is primarily used for models that rely on structural information, such as antibody-antigen complexes.
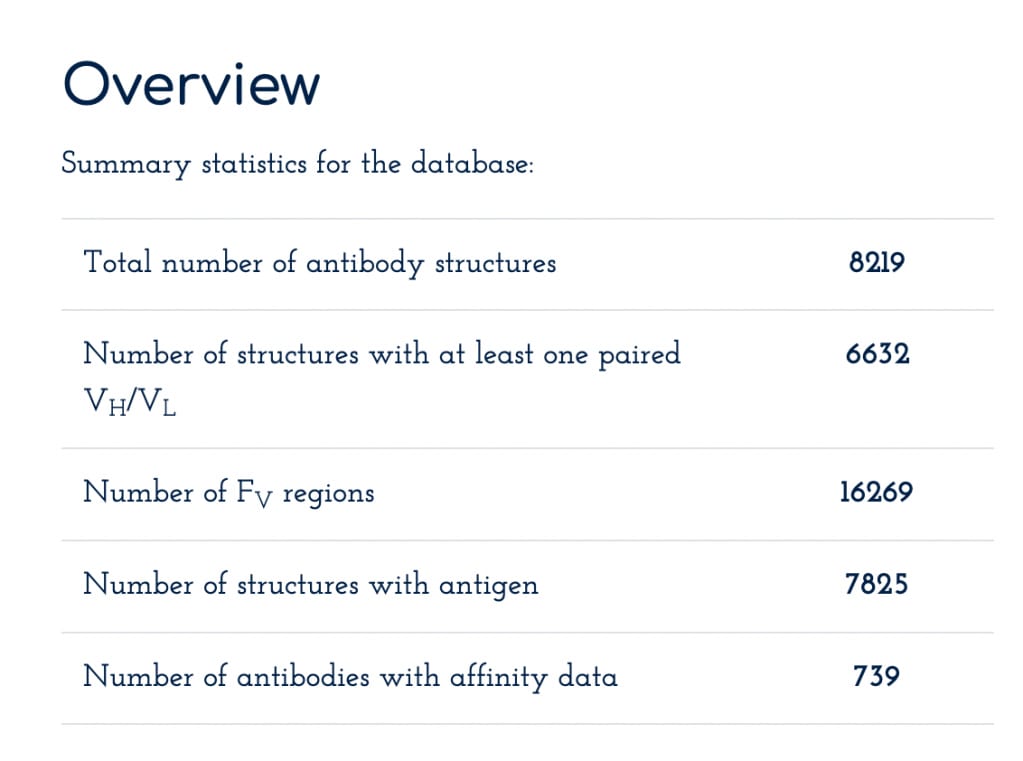
From here
Predicting antibody folds
The protein folding problem is a familiar one to any biologist and has been, in the monomer case, largely solved by Alphafold2. Being able to easily access the fold of any protein can explain a great deal about how a protein interacts with the world around it. Antibodies are no exception to this, the final fold of the paratope (remember, HCDR1-3 and LCDR1-3!) is the deciding factor in well it can bind to certain antigens. If we could accurately model antibody folds, the aforementioned molecular dynamic methods may become all that more powerful.
Interestingly, despite the variability of the CDR regions, the primary challenge of predicting the final antibody fold stems from HCDR3 alone! This is a repeating pattern in many of these papers, despite all CDR's being technically involved in antigen binding, HCDR3 is consistently the most important + challenging to modify. The paper has this to say about it:
Five of the CDR loops tend to adopt canonical folds that can be predicted effectively by sequence similarity. However, the third CDR loop of the heavy chain (CDR H3) has proven a challenge to model due to its increased diversity, both in sequence and length. Further, the position of the H3 loop at the interface between the heavy and light chains makes its conformation dependent on the inter-chain orientation. Given its central role in binding, advances in prediction of H3 loop structures are critical for understanding antibody-antigen interactions and enabling rational design of antibodies.
IgFold was an attempt to see if an LLM focused on only antibody folding could outperform Alphafold-Multimer, a version of Alphafold2 extended to work with multimers generally. Alphafold-Multimer was a step forwards for modeling the folding of multimeric proteins, but it wasn't quite the slam-dunk as the original model was for monomers, so IgFold did have some claim to make for the problem.
It was trained on 4,275 paired antibodies and nanobodies from SAbDab, but also had this (in my opinion, strange) second step of data augmentation where it used Alphafold2-predicted (not Alphafold-Multimer, because it hadn't been released yet...but it was released in time for the validation?) structures of 26,971 unpaired and 16,141 paired sequences from OAS.
The final result was only slightly positive; IgFold was largely identical to Alphafold-Multimer in accuracy (maybe slightly better). However, whereas Alphafold-Multimer takes over 10 minutes to run per antibody sequence, IgFold prediction was typically under 25 seconds; a 24x speedup. As far as I can tell though, these sorts of antibody structure prediction models haven't lead to anything particularly interesting in terms of antibody engineering.
LLM-guided antibody mutations
The basic idea here is to take FDA-approved antibodies that have already been through an optimization process and re-optimize them via proteomics LLM's. The way they did this was by computing log-likelihoods of every single-amino-acid-substitution of the VHC and VLC. So, for example, switching out a glycine to a cysteine on a VHC, and asking a set of pLM if the mutated version has 'higher likelihood to exist in nature'. These are questions that protein LLM's are quite good at answering, since they have been trained on a significant fraction of all proteins that have ever evolved. There were six LLM's used in total (ESM-1b and the five models part of ESM-1v), and they took the consensus of what all 6 said. They performed two rounds of optimization, the first round mutating a single residue and the second round mutating another one on top of the prior one. Importantly, no extra training of these networks were done, they used the pre-trained weights.
I originally learned about this paper from a Derek Lowe post, and his words best sum up the extraordinary results:
The results are impressive, particularly because they’re not starting with random antibodies to random proteins. No, they start with seven that are already in clinical use, and thus have already been through stages of optimization for their binding affinities and physical properties. Even with these, the pLM-suggested mutations are an improvement - better thermal stability, lower immunogenicity, and in every single case better potency in neutralization assays. This after scoring fewer [than] 20 suggested variants for each case and after two rounds of traditional laboratory evolution, which is far less work than usual for this kind of thing.
One minor nit about Derek's overview: he interprets the papers usage of pre-optimized antibodies as particularly impressive, but this is likely a far easier problem than optimizing random antibodies. It's a reasonably common experience in life-science ML to find that models are quite good at optimizing things that already have desirable properties, but terrible at finding those things in the first place. Similar phenomenons pop up elsewhere too, in binder generation, small-molecule generation, and so on. Of course, I cannot confirm this for sure, I just heavily suspect it to be the case.
A final interesting part of the study is that a majority of beneficial mutations were not in the CDR regions! For all I've been discussing the importance of paratope mutation, there may be a significant amount of value in exploiting mutations outside of it. From the paper:
Thirty-six out of all 76 language-model-recommended, single-residue substitutions (and 18 out of 32 substitutions that lead to improved affinity) occur in framework regions, which are generally less mutated during conventional affinity maturation compared to the complementarity-determining regions (CDRs)
De-novo CDR design conditioned on an antigen
Folding an antibody is really a step removed from the problem we really care about. What we'd really like is the ability to provide an antigen structure and have the model design an antibody specifically tailored to bind that antigen with high affinity and specificity. A paper in early 2023 poked at this exact problem, they designed fully antibodies that bind to a given antigen, all via using a model that has never seen antibodies that bind to that specific antigen! This is the 'de-novo' part, there are many antigens for which we've never observed an antibody that could bind to it, so a model that doesn't require that is exceptionally powerful.
They redesigned HCDR3 regions (HCDR3 coming up again as important!) of a well-known antibody (trastuzumab) that binds to an antigen protein called HER2. Given only the HER2 amino-acid sequence, they had the model fill in the HCDR3 regions of trastuzumab. What exactly is this 'model'? It is unfortunately not discussed at all in the paper, likely for commercial reasons given that an antibody design company released the paper.
Notably, this means we are running into the same issue as before with the LLM evolution paper! Given that binding outside of HCDR3 is known to be important and that trastuzumab contains mutations outside of HCDR3, we may expect that the modeling strategy here may pick a 'free ride' on the already good sequence space that the non-HCDR3 regions of trastuzumab inhabit. Moreover, is this actually 'de-novo', if we're starting from a validated antibody. We'll return to this point later on...
Via their model, they generate 400k possible antibodies and test binding of them in a high-throughput fashion to the HER2 antigen. Of these, 4k show possible binding and 421 of them were selected for follow-up validation via pre-filtering by some molecular dynamics magic they explain in the supplementary material. 71 of these had < 10nM Kd values (remember, <10nM Kd is the range of 'high' affinity), and three of them had higher Kd values than trastuzumab. They claim high sequence diversity amongst all generated antibodies. This is definitely a win for HCDR3 design in some capacity, though the issues raised above should be kept in mind.
Things are less rosy when the model is extended to redesign all HCDR regions...though, of course, while these fully redesigned antibody Kd values aren't terrible, they are quite far off from an established drug. The dream of full antibody design is far off, even redesigning HCDR regions (and not HCDR, or even non-CDR regions) of an established antibody is still quite challenging!
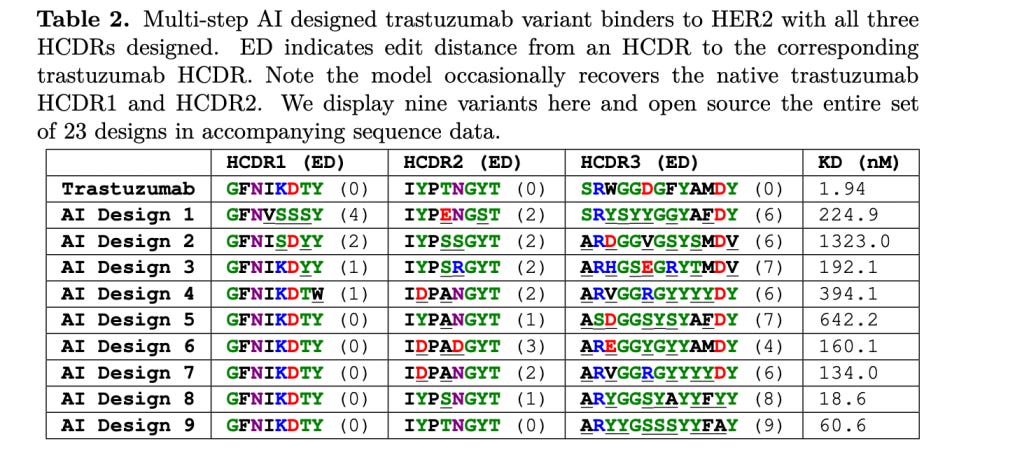
There's another reason we're discussing this paper, and thats because of an interesting twitter (X) thread that another antibody design startup made about this work! They were fairly pessimistic about the results and mirror our HCDR3 + de-novo concerns from earlier, and we're finally in a place to understand their critiques, so I highly recommend reading the thread! It's a good way to test our understanding of the subject:
https://twitter.com/SurgeBiswas/status/1613232556673224705?s=20&t=msepm203T3AoyEkIuMRaDA
So, what's the verdict here? There really isn't one, the role of ML in antibody design is still fuzzy and what is 'impressive' or worthy of attention is still a little vague. The company behind this paper did release some follow-up work that did directly address some of the concerns raised in the thread, using a method called 'IgDesign' that generates de-novo antibodies conditioned on an antigen. We won't discuss this paper heavily, since the results are, in my opinion, much more nuanced and difficult to explain without an entire post dedicated to it. But I do highly recommend reading the paper! The methodology involves redesign of HCDR1-3 (alongside HCDR3 only to compare) focused on a variety of antigens (still using a validated antibody for the antigen for the rest of the antibody though!), and they achieve some interesting results: For 5 out of 8 antigens (antigen 1, 2, 6, 7, and 8), IgDesign generates binders with equal or higher affinities to the reference antibody. For these 5 targets, several designed binders have affinities within one order of magnitude of the reference antibody’s affinity. If you're interested in what is the 'state of the art' when it comes to antibody design, I feel mildly confident in saying that it is this one.
De-novo nanobody design conditioned on an antigen
Of course the Baker lab, a legend in solving protein design challenges, has an approach to this. And it comes closest to our dream of complete design of an antibody with nothing more than the antigen provided as input. They use a re-trained RFDiffusion model as their primary method for doing this. Getting into the weeds of RFDiffusion would be really hard, so, while I do have a post of RFDiffusion that explains some very surface-level details of the model, we could simply view this re-trained RFDiffusion model as a magic box that produces new antibodies given an antigen.
But, it's not exactly an antibody that they are designing, it's a 'nanobody', or VHH. In the eyes of this paper, designing full antibodies isn't particularly important, given that smaller, more compact versions of those antibodies work with basically the same efficacy, and maybe have some advantages as well. We've discussed these form of antibodies earlier in this post, but as a reminder, nanobodies are composed of only one of the VHC's of an antibody; so, only 3 CDR's, all of them heavy, and no FWR region.
Let's walk through exactly what they did. Their model creates structures and sequences for HCDR1-3 regions, but, once again, keeps the FWR region provided by the user + constant. Moreover, unlike all prior method(s), they are able to target specific epitopes of an antigen at inference time for the nanobody to bind to, which is a huge step forwards in design. They use an established nanobody that has been confirmed to work in humans as the framework for all designs called 'NbBcII10FGLA', which is basically just a natural nanobody found in camels but humanized. Finally, they design nanobodies for a range of antigens: Clostridium difficile toxin B (TcdB), influenza H1 hemagglutinin (HA), respiratory syncytial virus (RSV) sites I and III, SARS-CoV-2 receptor binding domain (Covid RBD) and IL-7Rɑ, so, lots of diversity in antigens.
So, what are the results? Best for them to state it in their own terms:
9000 designed VHHs were screened against RSV site III and influenza hemagglutinin with yeast surface display, before soluble expression of the top hits in E. coli. Surface Plasmon Resonance (SPR) demonstrated that the highest affinity VHHs to RSV site III and Influenza Hemagglutinin bound their respective targets with 1.4μM and 78nM respectively. C) 9000 VHH designs were tested against SARS-CoV-2 receptor binding domain (RBD), and after soluble expression, SPR confirmed an affinity of 5.5μM to the target. Importantly, binding was to the expected epitope, confirmed by competition with a structurally confirmed de novo binder (AHB2, PDB: 7UHB). D) 95 VHH designs were tested against the C. difficile toxin TcdB. The highest affinity VHH bound with 262nM affinity, and also competed with an unpublished, structurally confirmed de novo binder to the same epitope (right)
So, while none of their designed nanobodies could be reasonably characterized as 'strong' binders to their antigen (nothing less than <10nM Kd), some could be described as moderate (<10μM Kd); there were also nanobodies that reached the <1 μM Kd region for HA and TcdB. Nanobodies for RBD and TcdB were also hitting the epitope desired during inference time, which is huge, especially considering that binders to this epitope weren't found in nature! They do little discussion on the diversity of sequences produced; I can't find any comparison to OAS, as is typically done to assess diversity, here.
Moderate results overall. A huge jump in applying antibody design techniques to a minimized form of antibody (which shows more promise than antibodies generally, even outside of AI), but the binding capabilities of these engineered nanobodies are lacking compared to what we expect from clinical-grade antibodies. However, this is closest to true de-novo design; the base nanobody used to scaffold the HCDR's was never explicitly meant to bind to any of the antigens, so a model being a 'freeloader' on non-HCDR changes is unlikely here. Hoping that the Baker lab releases a follow-up paper here soon given that RFDiffusion-AA was released, might dramatically improve accuracy.
Conclusion
Immunology is complicated, and antibody engineering using ML is still very much in the early days. I originally started this post expecting that there were some real magic bullets hiding away in papers I couldn't understand, but it seems like that isn't quite the case. People are still figuring out exactly what's going on in the field, and the results show that. There are aspects of ML-assisted antibody engineering that aren't even yet being discussed, like polyreactivity and, up until the Baker lab paper just a month ago, even epitope targeting! And I'm sure there are more nuanced aspects of antibody development that I'm not even aware of and are crucially important to rational antibody designers, but hasn't even been touched by ML researchers.
But, as with everything in this field, things can change overnight!
0 comments
Comments sorted by top scores.