Can o1-preview find major mistakes amongst 59 NeurIPS '24 MLSB papers?
post by Abhishaike Mahajan (abhishaike-mahajan) · 2024-12-18T14:21:03.661Z · LW · GW · 0 commentsThis is a link post for https://www.owlposting.com/p/can-o1-preview-find-major-mistakes
Contents
Introduction The 3 papers that o1 called out Protein sequence domain annotation using language models Guided multi-objective generative AI for structure-based drug design Loop-Diffusion: an equivariant diffusion model for designing and scoring protein loops Conclusion None No comments
TLDR: o1 flags major errors in 3 papers. Upon reviewing o1’s response, none of the papers have actual errors. However, it took the help of the authors to disprove o1’s criticism of the third paper (Loop-Diffusion), which was theoretically correct, but largely irrelevant for the problem the paper was studying. o1 probably shouldn’t be used to blindly review papers, but it does often have interesting thing to say.
Introduction
I recently saw this post on Twitter:
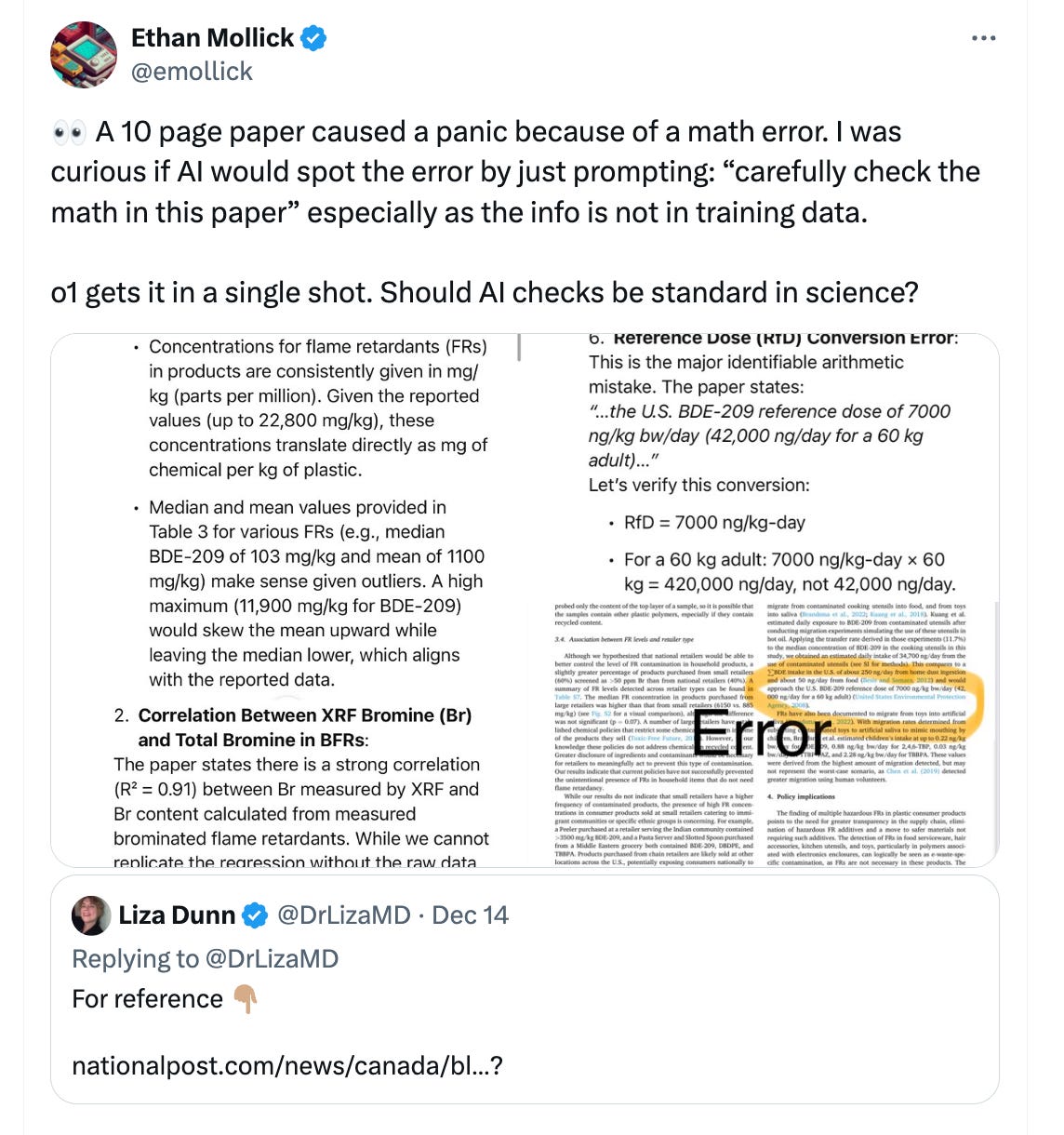
And got nerd-sniped by it.
I wondered if there could be anything similar done for the recent crop of work published at MLSB (Machine Learning in Structural Biology) workshop at this last NeurIPS 2024, just last week. I downloaded all papers from the workshop, of which there were 81. To my dismay, I discovered that a fair number of the links were broken, so I could only grab 59 of them.
I fed each one of them (through pure PDF → text conversion, which is obviously lossy, so keep that in mind) into the o1-preview API with the following prompt:
Assume the role of a deeply knowledgable reviewer, with a deep understanding of the fields of proteomics, chemistry, computational biology, and machine learning. Check the given paper for any major errors, either mathematical or biological in nature, that may invalidate the paper.
Keep in mind, these papers have already been published at NeurIPS, so if there are any errors, they are likely to be very subtle. Look very, very carefully. If there are any, please point them out. If there are none, please say so.
Dont bother with minor issues like formatting or small mistakes, or irrelevant errors that are in most ML papers (e.g. that the datasets aren't large enough).
A major error is something that is mathematically incorrect, or something that is biologically incorrect. It should be something that completely invalidates the conclusions of the paper.
To start off with, give a summary of the paper, ensuring that you cover all the concepts, ideas, and math used in the paper to check. Then, give a list of all the major errors you found in the paper, if there any. If there are none, say so.
Give all of this in the following format:
Summary:
Major Errors (if any):And saved the o1 response. From there on, I sifted through each of responses, and qualitatively decided whether o1 was ringing the alarm bell on major mistakes being present (for what its worth, o1 was pretty explicit). I did ignore any o1 screaming over future dates or future methods being present (e.g. Alphafold3, released in 2024), since its cutoff date is still in 2023.
From here, three papers were explicitly called out as having major errors. For each of the papers that were flagged, I’ll include o1’s response and I’ll try to give my best guess on whether the issue it called out is real or not.
This said, I’m obviously not an expert on everything! MLSB spans a lot of different fields (molecular dynamics, proteomics, cryo-EM, etc), and I’ve only actively studied a few of them. For each of my posts, I’ll also give some sense of how knowledgable I am about the area so you decide how much to trust me.
If curious, this all cost just about 40~ dollars. Not too bad!
The 3 papers that o1 called out
Protein sequence domain annotation using language models
How knowledgable I am about this?: Reasonably knowledgable.
My take: o1’s take is correct that the input sequences to this method (which relies on ESM2) may have homologous sequences in ESM2’s training set, which can be considered a form of data leakage. So it potentially hurts the main conclusion of the paper. But characterizing it as a ‘major error’ is a bit harsh. The authors ensured that the input sequences were not in Uniref50 (and thus outside of ESM2’s immediate training data), and that’s about all you can really do for these sorts of papers. Ideally, you retrain a pLM from scratch, but that feels a bit unnecessary. Most papers doing something similar are going to suffer from an identical limitation.
And, most importantly, the authors actively recognize this limitation! And devote a whole paragraph to it! From the paper: Despite our efforts to mitigate it, information from the test set may still contribute to training through the millions of sequences used to train ESM-2. While we exclude sequences used to train ESM-2 from our 0-20 PID test subset, indirect leakage through homology remains a possibility. Ideally, retraining ESM-2 from scratch on our training data would provide better insight into the out-of-distribution generalization capabilities of PSALM. We have not done this in the present work because of the compute demand for training a pLM like ESM-2, but we plan to rigorously split a much larger set of proteins to train a pLM from scratch.
Guided multi-objective generative AI for structure-based drug design
How knowledgable I am about this?: Somewhat knowledgable.
My take: I feel like o1 is technically right in one direction: Vina scores as a proxy for binding affinity isn’t great, so relying on it as a benchmark for how well your model is doing also isn’t great. But it’s also…a pretty common thing to use. Ideally there would’ve been experimental validation, but there wasn’t, and that’s not especially unusual from an ML small molecule paper.
There’s two more bizarre comment here.
One about how the authors shouldn’t have tried to predict synthetic accessibility from 3D structures because it’s traditionally calculated via 2D structures…which, like, okay? So? Two, there was a bit about unfair comparisons (virtual screening of a chemical library versus their method) which was initially a bit interesting, but it once again doesn’t make much sense. At worse, the authors didn’t optimize their virtual screening baseline, which again isn’t too out of the norm. What I had hoped o1 would mention is something similar to this inductive.bio post — which is about how docking baselines in ML papers are often badly done — but unfortunately it went a much less interesting direction.
Loop-Diffusion: an equivariant diffusion model for designing and scoring protein loops
How knowledgable I am about this?: Not very knowledgable.
My take: I don’t really understand how energy-based models work and my understanding of dynamics also isn’t great, so my comments here aren’t super valuable. My initial instinct was that even if the math error was real and you had varying additive energy constants between inputs, it still isn’t that big of a deal. A lot of things don’t matter when you’re working with energy differences (as opposed to absolute energies), and constants felt more tied to absolute energies, so o1 is wrong. But again, I don’t know much about this area.
I did check in with a coworker, Jakub, who knows much more about this area. His initial instinct was that the o1 response was wrong, but wasn’t 100% positive given how the paper discussed the constant.
Luckily, he also knew the authors of this paper, so he forwarded o1’s comments to them. Surprisingly, this led to a fair bit of discussion. As it turned out, o1 is somewhere in the realm of theoretically correct: additive constants can vary for any given point on an energy landscape if the system (defined below) dramatically changes, but for the problems under consideration in the paper, it is unlikely to vary significantly. From the authors:
o1 isn’t entirely wrong but it is a bit of a stretch to call this a major error. While the mentioned energy constant doesn’t depend on the loop conformation, it can in principle depend on the “system” i.e. the molecular structure of the protein (atom composition, bond connectivity, etc.). Such a constant would be a global property of the system, e.g. a function of the number and the type of atoms in the environment. It should also be noted that in our comparisons of different loop variants, the introduced substitutions typically result in addition or removal of only a handful of atoms within a local environment of a few hundred atoms. Consequently, we do not expect these alterations to significantly change the associated energy constants. This likely explains why our predicted differences in loop energies closely approximate the loop mutational effects on the binding affinities in T-cell receptors. These considerations are however important, and we are currently working to refine our preliminary model architecture to infer more physically motivated energies.
So, once again, o1 shouldn’t have flagged this paper.
Conclusion
Obviously, we don’t know the actual split-up of has-no-errors and has-errors amongst the 59 papers used here. And 59 is quite a small sample set! But it generally seems like, in at least one domain, o1 was not useful in identifying major issues. In all 3 cases where o1 made a fuss, o1 was either entirely wrong, or identified problems that ended up not being that big of a deal.
Still, it was interesting to read what o1 thought, especially for the third paper! I’ll note that, for fun, I actually ran the above papers through o1 again, and the third paper (Loop-Diffusion) was once again flagged (the other two weren’t)! But this time for a completely different error: an incorrect negative sign in one of their equations. Here is o1’s second response. Since I already had the authors ears, o1’s response was also sent to them. Once again, o1’s was wrong, but the authors did agree that the notation was slightly confusing and will be addressed in a followup paper:
As for the second comment, this is not an error and our calculations are consistent with the definitions provided in the manuscript. In our notation, the loss function is ||\epsilon_\theta+\epsilon||^2, which is consistent with the approach taken in Song et al for score matching criteria. This leads to the loss function that we present in equation 4. We chose this to be more consistent with our background and with downstream ddG prediction tasks, and we hoped that an interested human reader would pick up on this difference. It appears that o1 is simply comparing against the original DDPM paper without thinking about how the difference arises. Nonetheless, the comment by o1 highlights the confusion that our notation could cause (even for humans), and we will likely change out notation in the followup version of this manuscript to enhance clarity.
I’d consider reading through the o1 responses as a learning exercise, and a good way to grapple with the possible (emphasis on possible) failure modes of a paper. As always, models like these are going to get better, but it doesn’t seem like it’s quite all there right now for replacing real reviewers. Still, interesting overall!
0 comments
Comments sorted by top scores.