A primer on why computational predictive toxicology is hard
post by Abhishaike Mahajan (abhishaike-mahajan) · 2024-08-19T17:16:37.735Z · LW · GW · 2 commentsThis is a link post for https://www.owlposting.com/p/a-primer-on-why-computational-predictive
Contents
Introduction Some background The hard stuff The relevance of toxicity datasets to the clinical problem Methodological problems in toxicity datasets Intraspecies toxicity variability Toxicity synergism Conclusion None 2 comments
Introduction
There are now (claimed) foundation models for protein sequences, DNA sequences, RNA sequences, molecules, scRNA-seq, chromatin accessibility, pathology slides, medical images, electronic health records, and clinical free-text. It’s a dizzying rate of progress.
But there’s a few problems in biology that, interestingly enough, have evaded a similar level of ML progress, despite there seemingly being all the necessary conditions to achieve it.
Toxicology is one of those problems.
This isn’t a new insight, it was called out in one of Derek Lowe’s posts, where he said: There are no existing AI/ML systems that mitigate clinical failure risks due to target choice or toxicology. He also repeats it in a more recent post: ‘…the most badly needed improvements in drug discovery are in the exact areas that are most resistant to AI and machine learning techniques. By which I mean target selection and predictive toxicology.’ Pat Walters also goes into the subject with much more depth, emphasizing how difficult the whole field is.
As someone who isn’t familiar at all with the area of predictive toxicology, that immediately felt strange. Why such little progress? It can’t be that hard, right? Unlike drug development, where you’re trying to precisely hit some key molecular mechanism, assessing toxicity almost feels…brutish in nature. Something that’s as clear as day, easy to spot out with eyes, easier still to do with a computer trained to look for it. Of course, there will be some stragglers that leak through this filtering, but it should be minimal. Obviously a hard problem in its own right, but why isn’t it close to being solved?
What’s up with this field?
Some background
One may naturally assume that there is a well-established definition of toxicity, a standard blanket definition to delineate between things that are and aren’t toxic. While there are terms such as LD50, LC50, EC50, and IC50, used to explain the degree by which something is toxic, they are an immense oversimplification.
When we say a substance is "toxic," there’s usually a lot of follow-up questions. Is it toxic at any dose? Only above a certain threshold? Is it toxic for everyone, or just for certain susceptible individuals (as we’ll discuss later)? The relationship between dose and toxicity is not always linear, and can vary depending on the route of exposure, the duration of exposure, and individual susceptibility factors. A dose that causes no adverse effects when consumed orally might be highly toxic if inhaled or injected. And a dose that is well-tolerated with acute exposure might cause serious harm over longer periods of chronic exposure. The very definition of an "adverse effect" resulting from toxicity is not always clear-cut either. Some drug side effects, like mild nausea or headache, might be considered acceptable trade-offs for therapeutic benefit. But others, like liver failure or birth defects, would be considered unacceptable at any dose. This is particularly true when it comes to environmental chemicals, where the effects may be subtler and the exposure levels more variable. Is a chemical that causes a small decrease in IQ scores toxic? What about one that slightly increases the risk of cancer over a lifetime (20+ years)?
And this is one of the major problems with applying predicting toxicology at all — defining what is and isn’t toxic is hard! One may assume the FDA has clear stances on all these, but even they approach it on a ‘vibe-based’ perspective. They simply collate the data from in-vitro studies, animal studies, and human clinical trials, and arrive to an approval/no-approval conclusion that is, very often, at odds with some portion of the medical community.
Of course, we needn’t get extremely precise with what isn’t toxic or not toxic to start off with — something are painfully obviously toxic, whereas other things aren’t. One common method of handling toxicity earlier in the drug discovery process is to minimize the creation of ‘toxicophores’, or structural motifs in chemical designs that are known to cause downstream issues, during the design process, such as nitroaromatic compounds (a hyperbolic case). The existence of easily recognizable toxicophores spurned interest in establishing mappings between facets of a chemical structure and the physiological impact it had on organisms, leading to a field of study called ‘Quantitative Structure-Activity Relationship’, or QSAR.
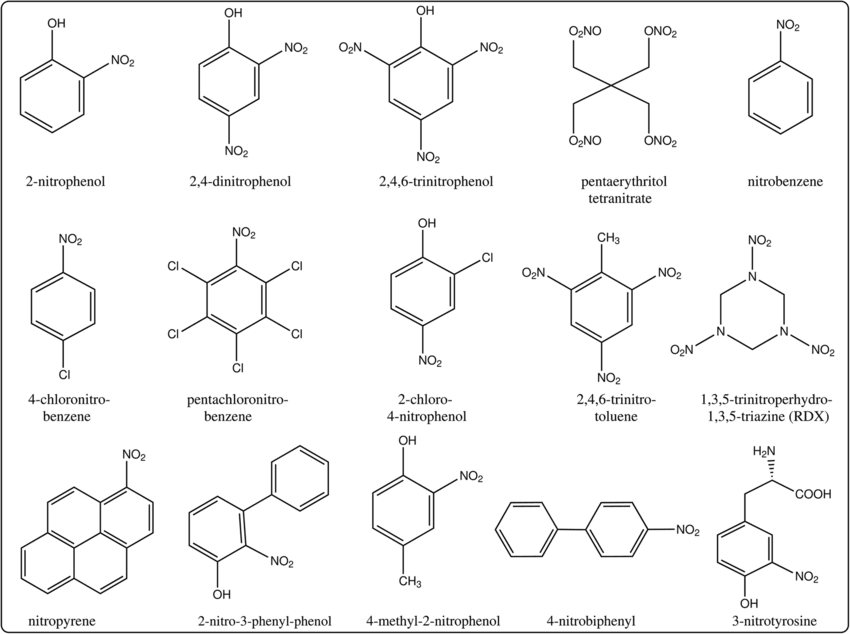
Nitroaromatic compounds. From here
Early forms of QSAR’s utilized hand-crafted features derived from a chemical structure, such as atom count, chemical bonds, and so on, as features to statistical models that learned their correlations to toxicity readouts (amongst other things). In time, the count of these chemical fingerprint features slowly grew, attempting to encompass every nuanced characteristic of a drug — eventually including measurements about how the chemical interacts with the world, such as their solubility in water or binding to certain enzymes. As with every other field, the explosion of deep learning led to a pivot — instead of working with derived features understandable to a chemist, neural networks were instead given the raw molecule as input, represented in either 2D or 3D space, building their own conception of what is/isn’t important for the problem of toxicity. But still, little massive progress. A recent (March 2024) Science paper applied transformers to the problem, walking away triumphant over more basic QSAR models, but no Alphafold-level jump in capabilities.
What’s missing?
The hard stuff
The relevance of toxicity datasets to the clinical problem
There’s a more fundamental problem here: the datasets we use to train predictive toxicology models are potentially too simplified for us to benefit from, even if models using them have perfect accuracy.
The Tox21 and ToxCast (both subsets from a larger dataset called MoleculeNet), are both very widely used datasets for predictive toxicology. They both contain dozens of different cellular assay readouts related to things like how drugs changed nuclear receptor activity, stress response pathways, and various cytotoxicity markers. But the biological relevance of many of these individual in-vitro assays to true organism toxicity is on shaky ground. One could say that any toxicity seen in-vitro will likely be seen in-vivo as well, but it’s unclear how true this is either. Cell lines may have unrealistically sensitive reactions to certain compounds, compounds may be toxic in petri-dishes but lose a fair bit of bioavailability upon ingestion, and the concentrations of drugs delivered via the blood stream may be dramatically lower than the ones given to cell lines. In-vitro is always a good start, but in-vivo translation must occur at some point!
The ClinTox dataset in MoleculeNet does attempt to touch on a more complex notion of toxicity via a label denoting whether an in-vivo clinical trial using a given drug found that it was toxic. But clinical toxicity here is boiled down to a 1/0, no notion of whether the drug displayed hepatotoxic, cardiotoxic, neurotoxic, or otherwise properties. Another similar dataset is TOXRIC, which annotates a wide range of molecules with in-vivo, in-vitro, and qualitative toxicity measurements, specifying whether drugs display acute toxicity, carcinogenetic properties, respiratory toxicity, and 12 other categories. But, while this goes far to include in more dense label information for each molecule, the underlying physiological impact of the toxicity is still missed!
But why is the underlying ‘toxicity phenotype’ important?
To answer this, I’d like to refer to the Stanford-released CheXpert dataset, a collection of 500,000~ chest x-ray’s with diagnostic annotations released back in 2019. It was the largest medical image dataset released at the time, but the clinical utility of any model built off it was questionable! There were a lot of issues with the dataset, one of the more interesting ones being that the human-performance accuracy rate was artificially low, since the X-ray had been sufficiently down-sampled enough from its original resolution such that some conditions became nearly impossible to detect. But the problem much more relevant to the toxicity discussion was the so-called hidden stratification problem; chest x-rays with a certain diagnosis label could be further subdivided into subtly different conditions with significantly different clinical outcomes. The last part is important, because otherwise the existence of a subclass underneath the labeled class isn’t actually useful for a model to be aware of. This exact situation may have a parallel in the toxicology dataset world; there is a whole world of hidden classes underneath the basic toxicity labels attached to each chemical and lacking it may lead you to the meaningfully wrong direction! Some forms of toxicity, despite being in the same ‘class’ of toxic, may have significantly different underlying phenotypes! For example, a drug that causes ocular toxicity via immune system overreaction is far easier to deal with than a drug that is just straight-up toxic to ocular cells — one requires simply immune suppressors to use it, the other requires rethinking the drug entirely.
One could imagine a world in which we have access to so much toxicity data that this problem ceases to matter — the model will figure it out. But, as it stands, ClinTox is composed of only 1478 molecules, Tox21 + ToxCast with 15,000~ molecule, and TOXRIC with 100k+ molecules (in total, many of which lack all labels) — a sizable number, but a far cry from NLP-level token sizes. Perhaps pushing dataset sizes up even more alleviates this problem, but it feels more likely that alternate directions should be explored.
How could we fix this? Instead of relying on our own fuzzy definitions of toxicity, we could perhaps instead defer it to a model capable of understanding phenotypes of toxicity more nuanced than ours could ever be. Microscopy foundation models, like Phenom-Beta by Recursion Pharmaceuticals, feels like a step in the right direction — perhaps the next generation of toxicology datasets are images of cell lines, or histology slides from a patient, subjected to a certain chemical, and such foundation models are used to understand them. After all, we do see morphological cell changes after application of toxic drugs! Maybe there’s even a time element, a new image 2, 8, 24, and so on hours after the application of the drug. Of course, the bull case here is that Recursion hasn’t billed their platforms utility for toxicity prediction, so perhaps this isn’t the right direction…
Methodological problems in toxicity datasets
Outside of the current set of toxicity datasets not being entirely connected to the problem of clinical toxicity, the datasets themselves have quality issues! This is a bit of a cop-out, but I’d honestly recommend reading Pat Walter’s post about this, it goes into much more detail than I ever could. But here’s the general TLDR for the problems with the datasets that many predictive toxicology papers rely on:
- Invalid chemical structures that can't be parsed by common cheminformatics tools
- Inconsistent stereochemistry and chemical representations
- Combining data from different sources without standardization
- Poorly defined training/test splits
- Data curation errors like duplicate structures with conflicting labels
- Assays with high rates of artifactual activities
+ some other points also addressed in this post! Again, excellent read, highly recommend.
Intraspecies toxicity variability
While most drugs are designed to hit specific molecular targets, there's still a huge potential for person-to-person differences in how they're absorbed, distributed, metabolized and excreted (ADME properties). This pharmacokinetic variability can lead to big differences in the actual tissue-level exposure to a drug for a given dose.
Genetic polymorphisms in drug metabolizing enzymes are the primary case of this phenomenon. For example, Cytochrome P450 2D6 enzymes are responsible for the metabolism of a huge number of drugs. The enzyme is encoded for by the CYP2D6 gene; the variations of which can lead to immense differences in drug clearance and bioavailability. For example, people with certain CYP2D6 polymorphisms are "poor metabolizers" of drugs like codeine and can end up with much higher exposure levels compared to the average person. There are also "ultra-rapid metabolizers", who clear drugs so quickly that they may not get a therapeutic effect at normal doses. And this doesn’t cleanly translate to “poor metabolizers should receive lower dosages of drugs” either, because the chemical in question matters! If the chemical is such that metabolization of it results in a weaker resulting chemical, the clinical impact of these polymorphisms will switch sides. And the rate of CYP2D6 variation isn’t particularly low either; one study pegged the rate of ultra-rapid metabolizers at 1-11% and poor metabolizers at 1-5% of the population, depending on the race. Finally, CYP2D6 isn’t even the only gene whose alleles can causes drug metabolism variation, there are way more — generally also known as ‘pharmacogenes’.
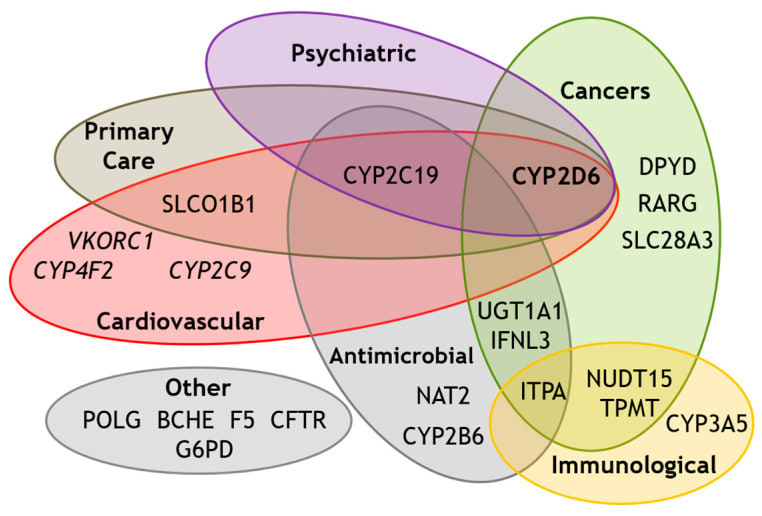
Known pharmacogenes. From here
What does this mean for ML? The very existence of pharmacogenes mean that any molecular-toxicity dataset that lacks sequence readouts of known pharmacogenes (and there may be unknown ones!) from the individual the data is derived from is, ultimately, limited in how generalizable it can be when applied to drugs for different individuals. Again, perhaps this problem eventually fixes itself with enough chemical data, but the case here is fishier. Even an all-powerful toxicology foundation model would be unable to pick up the underlying rules behind why drug toxicity variation exists if provided only toxic/not-toxic labels, it would simply model drug toxicity as a fundamentally noisy phenomenon.
How do we fix this? Full sequence readouts for every organism included in a toxicology dataset would obviously be prohibitively expensive. But there is a potential way out: real world evidence, or RWE. Those who have worked in RWE will understandably immediately recoil at this — it’s a field that is notorious for vastly overpromising and underdelivering, several blog posts could be written about how RWE datasets are rarely trustable + how companies leading the way in RWE have generally failed to capitalize on it. To be clear, I agree, but it’s still an interesting thought experiment!
RWE, often represented via insurance claims or electronic health records, was a big deal post-2015, or roughly when healthcare companies/national governments began to realize the potential value of the claims dataset they had. The core idea here was that, as a result of billing practices, we had accidentally created a low-fidelity dataset of an individual’s interaction with the healthcare system over their lifetime. We know their familial history, their chronic conditions, and so on, it’s all recorded somewhere. And perhaps, within it, is a similarly fuzzy representation of a patients set of pharmacogenes — indirectly represented within the joint distribution of the patients race, their conditions, their allergies, and everything else. If this sort of clinical data could be easily combined with toxicity datasets from phase 1/2/3 clinical trials, it may allow us to more deeply understand individual drug response heterogeneity, possibly helping us close this otherwise irreducible toxicology prediction error.
One last note: while pharmacogenes likely account for the majority of drug efficacy/toxicity, there is likely one more player: your microbiome. Very little has been published on the topic, but there are documented cases of gut flora affecting how a drug is metabolized! One major case is described here:
The dramatic impact of microbial metabolism on the toxicity of metabolites derived from drugs was clearly manifested in the death of fifteen patients, who were orally administered with sorivudine (SRV, 1-b-d-arabinofuranosyl-(E)-5-(2-bromovinyl) uracil) within forty days. This effect was attributed to the enterobacteria-mediated SRV hydrolysis, thus leading to the formation of 5-(2-bromovinyl) uracil. This transformation is mainly carried out by E. coli and Bacteroides spp. (B. vulgatus, B. thetaiotaomicron, B. fragilis, B. uniformis and B. eggerthii) and increases toxicity of the anticancer chemotherapy with 5-fluorouracil pro-drugs.
Toxicity synergism
Our final challenging problem are drug-drug interactions, also known as DDI. Drugs, especially amongst its largest consumers, do not exist in a vacuum; a fair bit of the US is on multiple drugs at the same time. And these drugs do interact in the bloodstream, potentially causing fatal events. An example of this phenomenon is with warfarin and aspirin — both extremely common drugs! If they are taken together, they will compete for binding to blood plasma proteins; the warfarin that cannot be bounded to plasma proteins will remain in the blood, eventually causing acute bleeding in patients. The rate of polypharmacy, which is taking five or more medications at a time, is between 10% and 50% depending on the age group. And to be clear, the warfarin-aspirin problem as described above isn’t exactly an edge case, one study found that amongst a patient population defined as having polypharmacy, the rate of at least one severe adverse effects from DDI were as high at 77%.
The complexity of predicting toxicity in these cases (maybe!) ramps up dramatically; it is likely that a fair number of such patients will have a drug regimen that’s largely unique to them alone. And the impact of pharmacogenes still exist, potentially even amplifying!
The state of the art is a bit fuzzy here. There has been headway in predicting DDI’s, but the datasets here are usually quite small in terms of number of molecules, on the order of a few hundred, often with many potential interactions missing (and subsequently being, maybe falsely, labeled as a negative example). And, given how common DDI’s are, it feels unlikely there is a current, good solution for it being done in drug-design beyond simple ‘does it interact with the same hypothesized mechanism’. It’s challenging to know the progress here; production-grade datasets here are, in my opinion, quite a long way off. This is true of many interaction-based problems in the life sciences and it’s especially true with toxicity-related datapoints.
It’s challenging to know how to fix this. But it may end up being a non-issue. Interactions between molecules in our body aren’t exactly orthogonal to the interactions between molecules and the body; everything is still atoms at the end of the day after all. Perhaps as we amass more singular molecular datapoints, we’ll accidentally get better at predicting DDI's. A similar phenomenon was seen with Alphafold2 in a mild sense; despite never having been trained on multimeric proteins, its monomer training regimen was enough such that it still performed well in the multimer case — though, of course, still worse than a version of Alphafold2 trained on multimers. But there’s an even more interesting possibility here: ultra-precise, high-throughput in-vivo screening. Gordian Biotechnologies Mosiac Screening platform feels immensely interesting in this regard. Their platforms allow one to use barcoded viruses to deliver drugs to extremely specific cells in-vivo, allowing you to test an incredibly high number of drugs in-vivo at the same time. With the current aim of the platform, it seems like these deliveries are meant to be to separate cells, ensuring that each drug can be understood independently of others. But one could imagine the platform be repurposed; perhaps multiple drugs could be delivered to the same set of cells, with thousands of different combinations, allowing us to create a large and high-fidelity drug interaction dataset extremely quickly. This said, the platform doesn’t currently bill itself as being able to better understand DDI’s, but more focused on the target discovery problem by speeding up in-vivo testing.
Conclusion
I really did scratch the surface of toxicology here, there’s so much material here. I am once again astonished by the immense amount of work on drug design written by medicinal chemists and biologists, and how little we still understand everything. I want to emphasize that toxicity is a really big deal. Each drug failing a clinical trial account for billions of wasted dollars and many thousands of work hours lost, and that rate of failure due to toxicity is frightingly high. One study has this to say about it:
Overall, approximately 89% of novel drugs fail human clinical trials, with approximately one-half of those failures due to unanticipated human toxicity
Even more concerningly, the danger of toxicity can remain danger even after approval, implying even a clinical trial isn’t the end-all-be-all for toxicity concerns. The same study continues:
Of 578 discontinued and withdrawn drugs in Europe and the United States, almost one-half were withdrawn or discontinued in post-approval actions due toxicity. Van Meer et al. found that of 93 post-marketing serious adverse outcomes, only 19% were identified in preclinical animal studies. In the first decade of the 21st century, approximately one-third of FDA-approved drugs were subsequently cited for safety or toxicity issues. or a combination of both, including human cardiovascular toxicity and brain damage, after remaining on the market for a median of 4.2 years
Despite all the problems we discussed here, I still believe the future is bright! There are so many scale-related things going on in biology right now, and it does feel like we’re hitting the precipice of something really interesting here.
2 comments
Comments sorted by top scores.
comment by Ponder Stibbons · 2024-08-20T14:20:46.194Z · LW(p) · GW(p)
A lot of what you write is to the point and very valid. However, I think you are missing part of the story. Let’s start with
“Unlike drug development, where you’re trying to precisely hit some key molecular mechanism, assessing toxicity almost feels…brutish in nature”
I assume you don’t really believe this. Toxicity is often exactly about precisely hitting some key molecular mechanism. A mechanism that you may have no idea your chemistry is going to hit before hand. A mechanism moreover that you cannot use a straight forward ML to find because your chemistry is not in any training set that an ML model could access. It is very easy to underestimate the vastness of drug-like chemical space, and it is generally the case any given biological target molecule (desired or undesired) can be inhibited or otherwise interfered with a wide range of different chemical moieties (thus keeping medicinal chemists very well employed, and patent lawyers busy). There is unlikely to be toxicological data on any of them unless the target is quite old and there is publically available data on some clinical candidates.
We look to AlphaFold as the great success for ML in the biological chemistry field, and so we should, but we need to remember that AlphaFold is working on an extremely small portion of chemical space, not much more than that covered by the 20 natural amino acids. So AlphaFold’s predictions can be comfortably within distribution of what is already established by structural biology. ML models for toxicology, on the other hand, are very frequently predicting out of distribution.
In point of fact the most promising routes to avoiding toxicity reside in models that are wholly or partially physics-based. If we are targeting a particular kinase (say) we can create models (using AlphaFold if necessary) of all the most important kinases we don’t want to hit and, using physics-based modelling, predict whether we could get unwanted activity against any of these targets. We still have the problem of hitting unrelated protein targets but even here we could, in principle, screen for similarities in binding cavities over a wide range of off-targets and use physics-based modelling to assess cases where there is a close enough match.
Needless to say that requires an awful lot of compute and no-one is really doing this to scale yet. It is a very difficult problem.
↑ comment by ChristianKl · 2024-08-21T22:57:48.583Z · LW(p) · GW(p)
I would also expect that the road is through creating models that predict off-target interactions. AlphaFold3 seems to be able to make some predictions about whether or not a given drug will bind with a particular proteins. Those aren't yet 100 percent accurate but Isomorphic Laboratories probably already does this kind of modeling.
Knowing off-targets itself doesn't tell you how exactly how serious the side-effects from hitting those off-targets happen to be but it's quite useful and allows avoiding the most egregious drug candidates that hit the most of targets.