I'm a bit skeptical of AlphaFold 3
post by Oleg Trott (oleg-trott) · 2024-06-25T00:04:41.274Z · LW · GW · 14 commentsContents
14 comments
(also on https://olegtrott.substack.com)
So this happened: DeepMind (with 48 authors, including a new member of the British nobility) decided to compete with me. Or rather, with some of my work from 10+ years ago.
Apparently, AlphaFold 3 can now predict how a given drug-like molecule will bind to its target protein. And it does so better than AutoDock Vina (the most cited molecular docking program, which I built at Scripps Research):
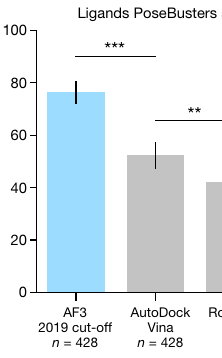
On top of this, it doesn’t even need a 3D structure of the target. It predicts it too!
But I’m a bit skeptical. I’ll try to explain why.
Consider a hypothetical scientific dataset where all data is duplicated: Perhaps the scientists had trust issues and tried to check each others’ work. Suppose you split this data randomly into training and test subsets at a ratio of 3-to-1, as is often done:

Now, if all your “learning” algorithm does is memorize the training data, it will be very easy for it to do well on 75% of the test data, because 75% of the test data will have copies in the training data.
Scientists mistrusting each other are only one source of data redundancy, by the way. Different proteins can also be related to each other. Even when the sequence similarity between two proteins is low, because of evolutionary pressures, this similarity tends to be concentrated where it matters, which is the binding site.
Lastly, scientists typically don’t just take random proteins and random drug-like molecules, and try to determine their combined structures. Oftentimes, they take baby steps, choosing to study drug-like molecules similar to the ones already discovered for the same or related targets.
So there can be lots of redundancy and near-redundancy in the public 3D data of drug-like molecules and proteins bound together.
Long ago, when I was a PhD student at Columbia, I trained a neural network to predict protein flexibility. The dataset I had was tiny, but it had interrelated proteins already:
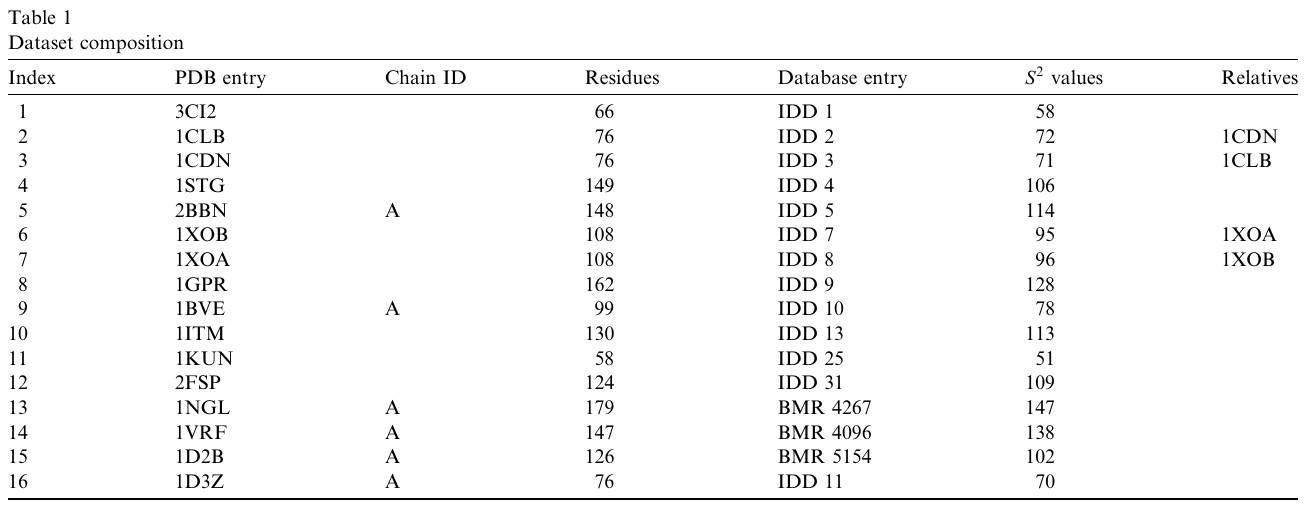
With a larger dataset, due to the Birthday Paradox, the interrelatedness would have probably been a much bigger concern.
Back then, I decided that using a random train-test split would have been wrong. So I made sure that related proteins were never in both “train” and “test” subsets at the same time. With my model, I was essentially saying “Give me a protein, and (even) if it’s unrelated to the ones in my training data, I can predict …”
The authors don’t seem to do that. Their analysis reports that most of the proteins in the test dataset had kin in the training dataset with sequence identity in the 95-100 range. Some had sequence identity below 30, but I wonder if this should really be called “low”:
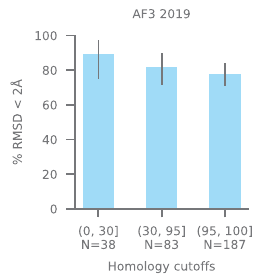
This makes it hard to interpret. Maybe the results tell us something about the model’s ability to learn how molecules interact. Or maybe they tell us something about the redundancy of 3D data that people tend to deposit? Or some combination?
Docking software is used to scan millions and billions of drug-like molecules looking for new potential binders. So it needs to be able to generalize, rather than just memorize.
But the following bit makes me really uneasy. The authors say:
The second class of stereochemical violations is a tendency of the model to occasionally produce overlapping (clashing) atoms in the predictions. This sometimes manifests as extreme violations in homomers in which entire chains have been observed to overlap (Fig. 5e).
If AlphaFold 3 is actually learning any non-obvious insights from data, about how molecules interact, why is it missing possibly the most obvious one of them all, which is that interpenetrating atoms are bad?
On the other hand, if most of what it does is memorize and regurgitate data (when it can), this would explain such failures coupled with seemingly spectacular performance.
14 comments
Comments sorted by top scores.
comment by Vanessa Kosoy (vanessa-kosoy) · 2024-06-25T04:59:37.900Z · LW(p) · GW(p)
This sounds like valid criticism, but also, isn't the task of understanding which proteins/ligands are similar enough to each other to bind in the same way non-trivial in itself? If so, exploiting such similarities would require the model to do something substantially more sophisticated than just memorizing?
Replies from: oleg-trott↑ comment by Oleg Trott (oleg-trott) · 2024-06-25T08:35:17.217Z · LW(p) · GW(p)
(ligand = drug-like molecule, for anyone else reading)
Right, I didn't mean exact bitwise memory comparisons.
The dataset is redundant(ish), simply as an artifact of how it's constructed:
For example, if people know that X binds A, and X ≈ Y, and A ≈ B, they'll try to add X+B, Y+A and Y+B to the dataset also.
And this makes similarity-based predictions look artificially much more useful than they actually are, because in the "real world", you will need to make predictions about dissimilar molecules from some collection.
I hope this makes sense.
comment by wassname · 2024-06-28T21:57:37.960Z · LW(p) · GW(p)
But if it's a poor split, wouldn't it also favour the baseline (your software). But they did beat the baseline. So if your concern is correct, they did outperform the baseline, but just didn't realistically measure generalisation to radically different structures.
So it's not fair to say 'it's only memorisation'. It seems fairer to say 'it doesn't generalise enough to be docking software, and this is not obvious at first due to a poor choice of train test split'.
comment by Ponder Stibbons · 2024-06-26T18:39:46.330Z · LW(p) · GW(p)
I have similar concerns regarding the ligand sets used to test Alphafold3. I’ve had a cursory look at them and it seemed to me there were a lot phosphate containing molecules, a fair few sugars, and also some biochemical co-factors. I haven’t done a detailed analysis, so some caveats. But if true, there are two points here. Firstly there will be a lot of excellent crystallographic training material available on these essentially biochemical entities, so AlphaFold3 is more likely to get these particular ones right. Secondly, these are not drug-like molecules and docking programs are generally parameterized to dock drug-like molecules correctly, so are likely to have a lower success rate on these structures than on drug-like molecules.
I think a more in-depth analysis of performance of AF3 on the validation data is required, as the OP suggests. The problem here is that biochemical chemical space, which is very well represented by experimental 3D structure, is much smaller than potential drug-like chemical space, which is poorly represented by experimental 3D structure comparatively speaking. So inevitably AF3 will often be operating beyond the zone of applicability, for any new drug series. There are ways of getting round this data restriction, including creating physics compliant hybrid models (and thereby avoiding clashing atoms). I’d be very surprised if such approaches are not currently being pursued.
Replies from: oleg-trott↑ comment by Oleg Trott (oleg-trott) · 2024-06-26T22:38:59.370Z · LW(p) · GW(p)
I'm surprised by how knowledgeable people are about this on this site!
BTW, there's some discussion of this happening on the CCL mailing list (limited to professionals in relevant fields) if you are interested.
comment by Yair Halberstadt (yair-halberstadt) · 2024-06-25T12:58:19.387Z · LW(p) · GW(p)
This makes sense, but isn't alphafold available for use? Is it possible to verify this one way or another experimentally?
Replies from: oleg-trott↑ comment by Oleg Trott (oleg-trott) · 2024-06-25T17:33:47.738Z · LW(p) · GW(p)
Determining 3D structures is expensive.
The most realistic thing one could do is repeat this work, with the same settings, but using k-fold cross-validation, where test and training sets are never related (like what I did at Columbia).
This will show how well (or poorly, as the case may be) the method generalizes to unrelated proteins.
I hope someone does it.
Replies from: ChristianKl↑ comment by ChristianKl · 2024-06-26T10:35:26.395Z · LW(p) · GW(p)
But do you need 3D structures to test alphafold? If alphafold makes a prediction about whether a given ligand binds to a protein, I would expect that testing whether or not that ligand binds to the protein is much cheaper.
Replies from: oleg-trott↑ comment by Oleg Trott (oleg-trott) · 2024-06-26T16:40:04.278Z · LW(p) · GW(p)
Unlike Vina, AF3 only predicts 3D structures, I believe. It does not predict binding affinities.
Replies from: ChristianKl↑ comment by ChristianKl · 2024-06-26T19:01:32.477Z · LW(p) · GW(p)
AlphaFold 2 was only predicting 3D structures. From the abstract of the Alpha Fold 3 paper:
far greater accuracy for protein–ligand interactions compared with state-of-the-art docking tools
The FastCompany article says:
Replies from: oleg-trottHe said that for the interactions of proteins with small (drug-like) molecules AlphaFold 3 showed 76% accuracy in a benchmark test (versus 52% for the next best tool).
↑ comment by Oleg Trott (oleg-trott) · 2024-06-26T19:16:20.588Z · LW(p) · GW(p)
Right. The benchmark (their test set) just compares 3D structures.
Side note: 52% also seems low for Vina, but I haven't looked into this further. Maybe the benchmark is hard, or maybe the "search space" (user-specified) was too big.
On their other test (in their Extended Data), both Vina and AF3 do much better.
comment by CuriousEcho · 2024-06-26T01:20:11.194Z · LW(p) · GW(p)
Since this shows up in homomeric proteins, maybe the issue is because symmetry breaking in quartinary structure happens late in training. This is just a guess tho... but aligns with my intuitions on training dynamics. I would also guess that this would get ironed out in the tails of the loss curve, if we push out further in training. I think the PLDDT loss would first learn to stack homomers on top of one another and then spread them out. I haven't used alphafold too much but the smoking gun plot id be interested in is clashes in heteromers vs clashes in homomers. It's a curious little detail, thanks for sharing this finding!
comment by Ponder Stibbons · 2024-06-27T11:32:58.773Z · LW(p) · GW(p)
There is an additional important point that needs to be made. Alphafold3 is using predominantly “positive” data. By this I mean the training data encapsulates considerable knowledge of favourable atom-atom or group-group interactions and relative propensities can be deduced. But “negative” data, in other words repulsive electrostatic or Van der Waals interactions, are only encoded by absence because these are naturally not often found in stable biochemical systems. There are no relative propensities available for these interactions. So AF3 can be expected to not perform as well when applied to real-world drug design problems where such interactions have to be taken into account and balanced against each other and against favourable interactions. Again, this issue can be mitigated by creating hybrid physics compliant models.
It is worth also noting that ligand docking is not generally considered a high accuracy technique and, these days is often used to 1st pass screen large molecular databases. The hits from docking are then further assessed using an accurate physics-based method such as Free Energy Perturbation.
comment by Tim Bonzon · 2024-06-25T23:54:48.270Z · LW(p) · GW(p)
So without knowing too much about AlphaFold 3 or AutoDock Vina, does this mean AlphaFold 3 essentially replaces AutoDock Vina? Does AutoDock Vina still fill a niche AlphaFold 3 doesn't?