What is “protein folding”? A brief explanation
post by jasoncrawford · 2020-12-01T02:46:09.003Z · LW · GW · 8 commentsThis is a link post for https://rootsofprogress.org/alphafold-protein-folding-explainer
Contents
9 comments
Today Google DeepMind announced that their deep learning system AlphaFold has achieved unprecedented levels of accuracy on the “protein folding problem”, a grand challenge problem in computational biochemistry.
What is this problem, and why is it hard?
I spent a couple years on this problem in a junior role in the early days of D. E. Shaw Research, so it’s close to my heart. Here’s a five-minute explainer.
Proteins are long chains of amino acids. Your DNA encodes these sequences, and RNA helps manufacture proteins according to this genetic blueprint. Proteins are synthesized as linear chains, but they don’t stay that way. They fold up in complex, globular shapes:
{kind=link}
One part of the chain might coil up into a tight spiral called an α-helix. Another part might fold back and forth on itself to create a wide, flat piece called a β-sheet:
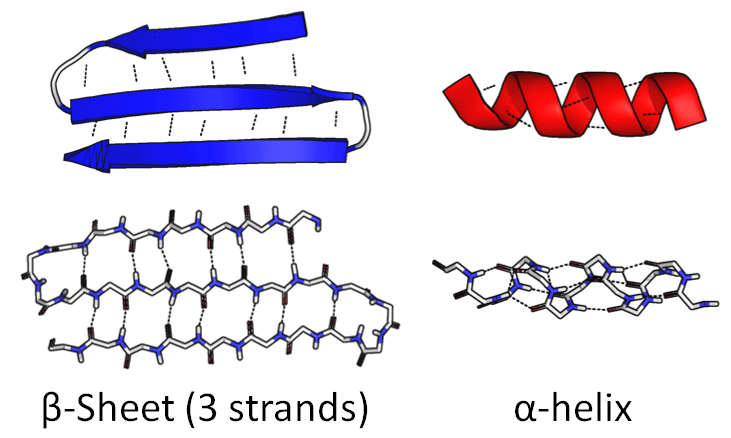
.png){kind=link}
The sequence of amino acids itself is called primary structure. Components like this are called secondary structure.
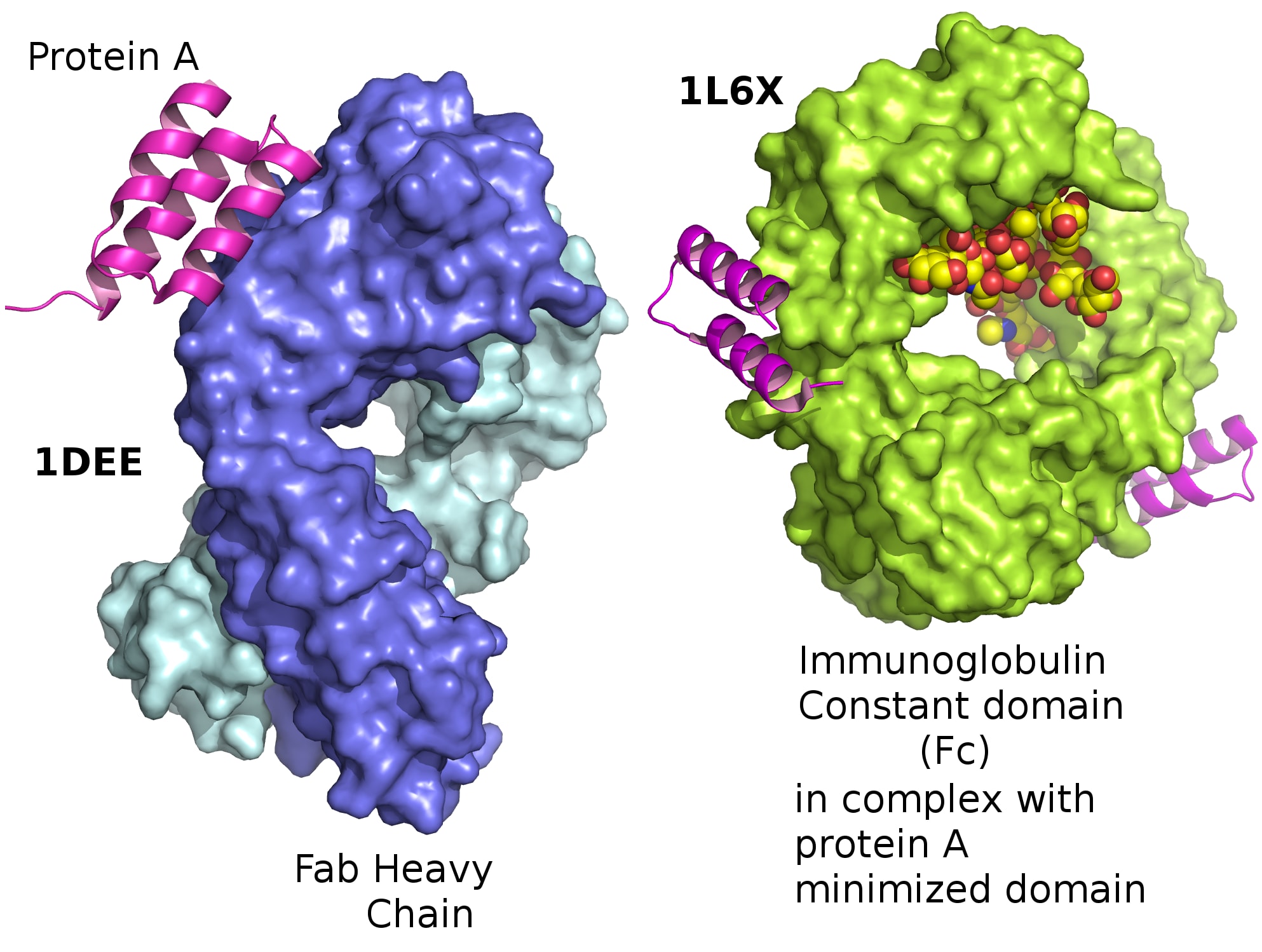
Then, these components themselves fold up among themselves to create unique, complex shapes. This is called tertiary structure:
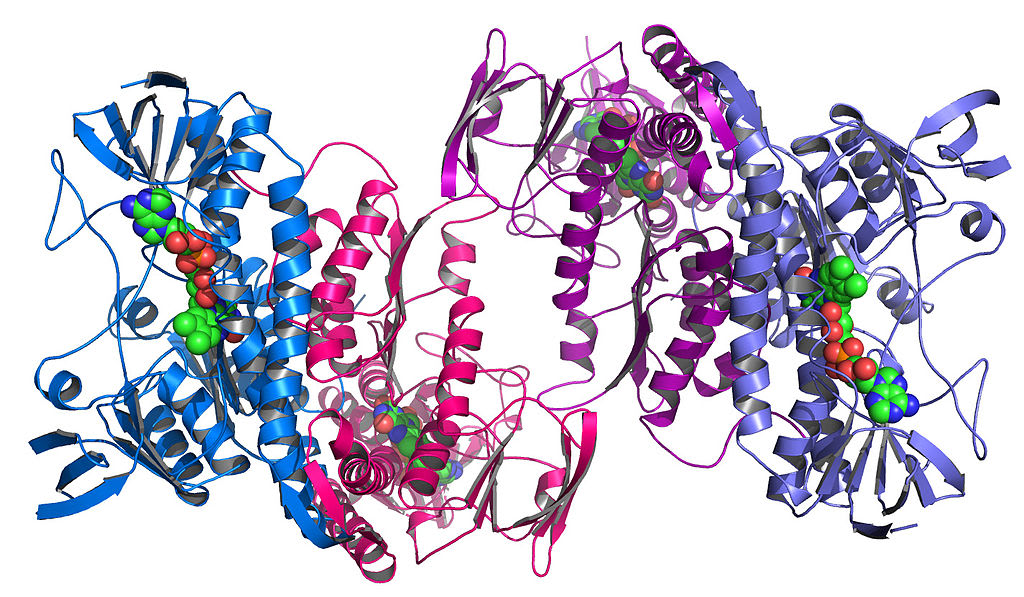
{kind=link}
This looks like a mess. Why does this big tangle of amino acids matter?
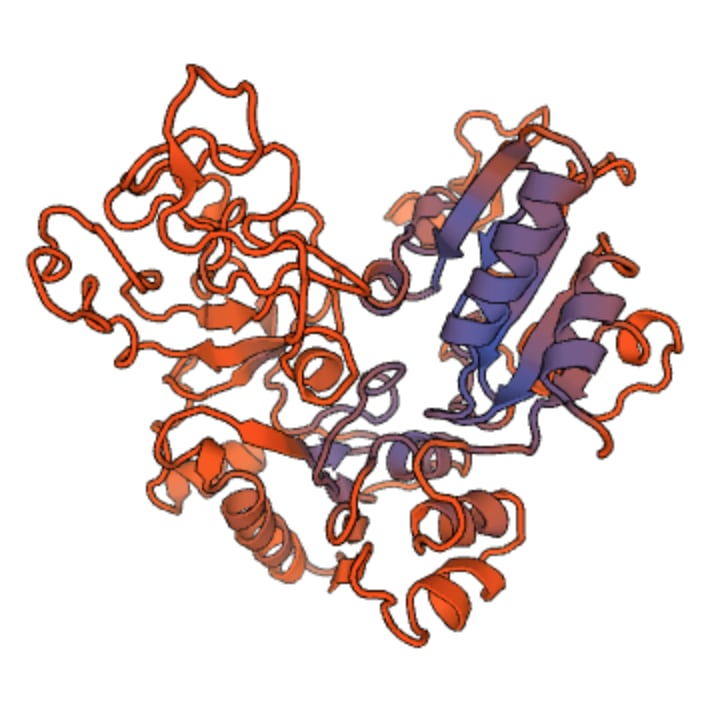
Protein structure is not random! Each protein folds in a specific, unique, and largely predictable way that is essential to its function. The physical shape of a protein gives it a good fit to targets it might bind with. Other physical properties matter too, especially the distribution of electrical charge within the protein, as shown here (positive charge in blue, negative in red):
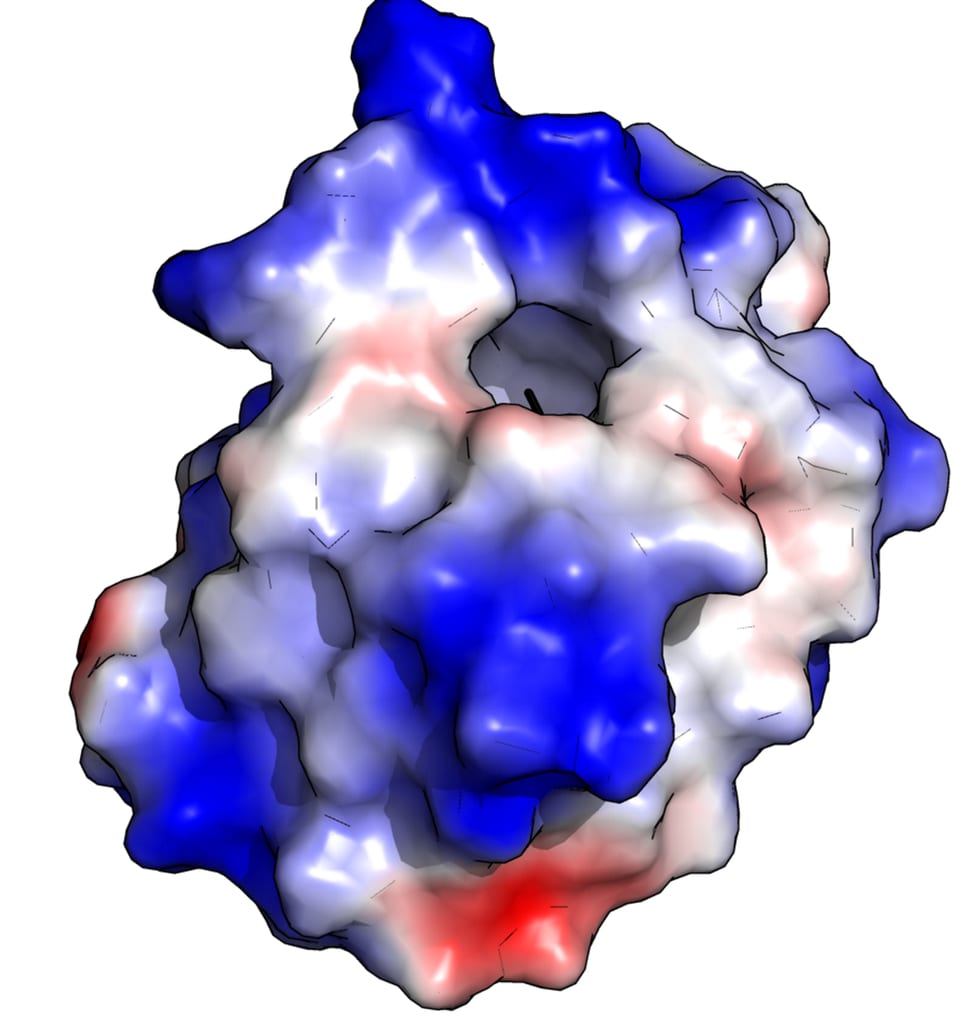
{kind=link}
If a protein is essentially a self-assembling nanomachine, then the main purpose of the amino acid sequence is to produce the unique shape, charge distribution, etc. that determines the protein’s function. (How exactly this happens, in the body, is still not fully understood, and is an active area of research.)
In any case, understanding structure is crucial to understanding function. But the DNA sequence only gives us the primary structure of a protein. How can we learn its secondary and tertiary structure—the exact shape of the blob?
This problem is called “protein structure determination”, and there are two basic approaches: measurement and prediction.
Experimental methods can measure protein structure. But it isn’t easy: an optical microscope can’t resolve the structures. For a long time, X-ray crystallography was the main method. Nuclear magnetic resonance (NMR) has also been used, and more recently, a technique called cryogenic electron microscopy (cryo-EM).

{kind=link}
But these methods are difficult, expensive, and time-consuming, and they don’t work for all proteins. Notably, proteins embedded in the cell membrane—such as the ACE2 receptor that COVID-19 binds to—fold in the lipid bilayer of the cell and are difficult to crystallize.
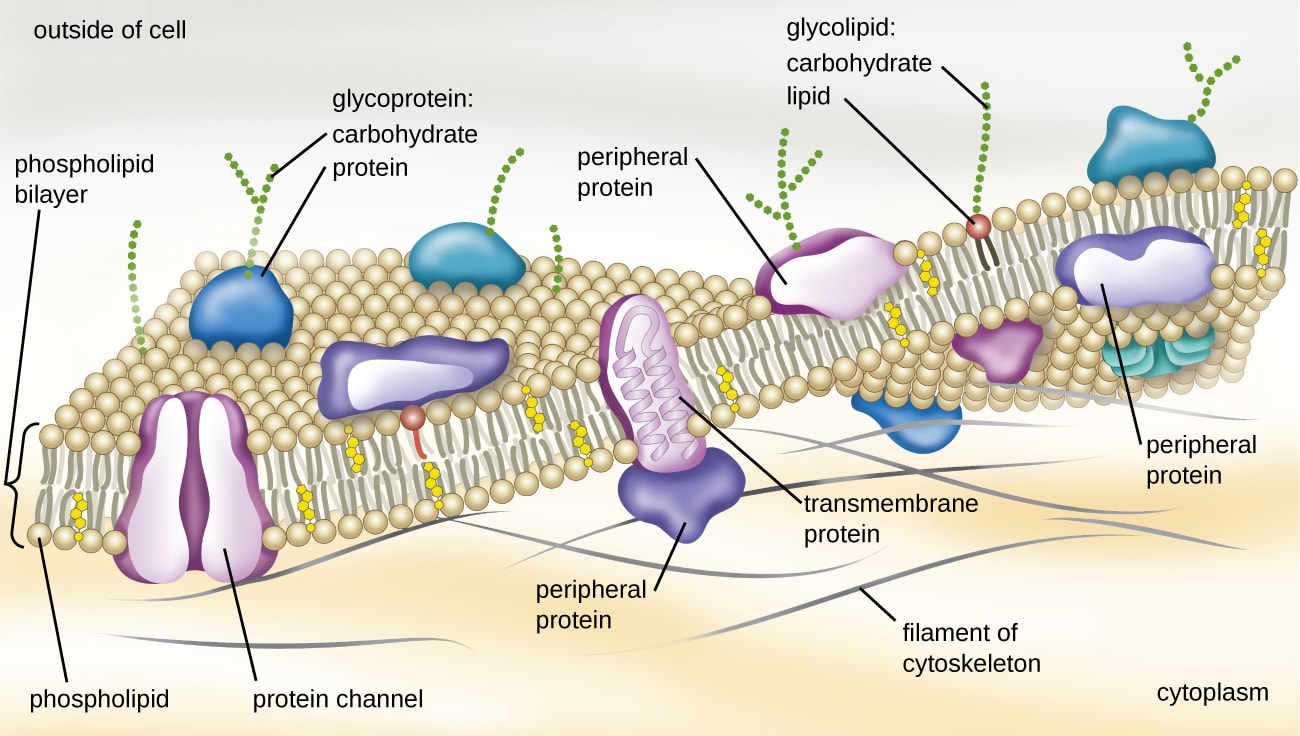
{kind=link}
Because of this, we have only determined the structure of a tiny percentage of the proteins that we’ve sequenced. Google notes that there are 180M protein sequences in the Universal Protein database, but only ~170k structures in the Protein Data Bank.
We need a better method.
Remember, though, that the secondary and tertiary structures are mostly a function of the primary structure, which we know from genetic sequencing. What if, instead of measuring a protein’s structure, we could predict it?
This is “protein structure prediction”, or colloquially, the “protein folding problem,” and computational biochemists have been working on it for decades.
How could we approach this?
The obvious way is to directly simulate the physics. Model the forces on each atom, given its location, charge, and chemical bonds. Calculate accelerations and velocities based on that, and evolve the system step by step. This is called “molecular dynamics” (MD).
The problem is that this is extremely computationally intensive. A typical protein has hundreds of amino acids, which means thousands of atoms. But the environment also matters: the protein interacts with surrounding water when folding. So you have more like 30k atoms to simulate. And there are electrostatic interactions between every pair of atoms, so naively that’s ~450M pairs, an O(N2) problem. (There are smart algorithms to make this O(N log N).) Also, as I recall, you end up needing to run for something like 109 to 1012 timesteps. It’s a pain.
OK, but we don’t have to simulate the entire folding process. Another approach is to find the structure that minimizes potential energy. Objects tend to come to rest at energy minima, so this is a good heuristic. The same model that gives us forces for MD can calculate energy. With this approach, we can try a whole bunch of candidate structures and pick the one with lowest energy. The problem, of course, is where do you get the structures from? There are just way too many—molecular biologist Cyrus Levinthal estimated 10300 (!) Of course, you can be much smarter than trying all of them at random. But there are still too many.
So there have been many attempts to get faster at doing these kinds of calculations. Anton, the supercomputer from D. E. Shaw Research, used specialized hardware—a custom integrated circuit. IBM also has a computational bio supercomputer, Blue Gene. Stanford created Folding@Home to leverage the massively distributed power of ordinary home computers. The Foldit project from UW makes folding a game, to augment computation with human intuition.
Still, for a long time, no technique was able to predict a wide variety of protein structures with high accuracy. A biannual competition called CASP, which compares algorithms against experimentally measured structures, saw top scores of 30–40%… until recently:
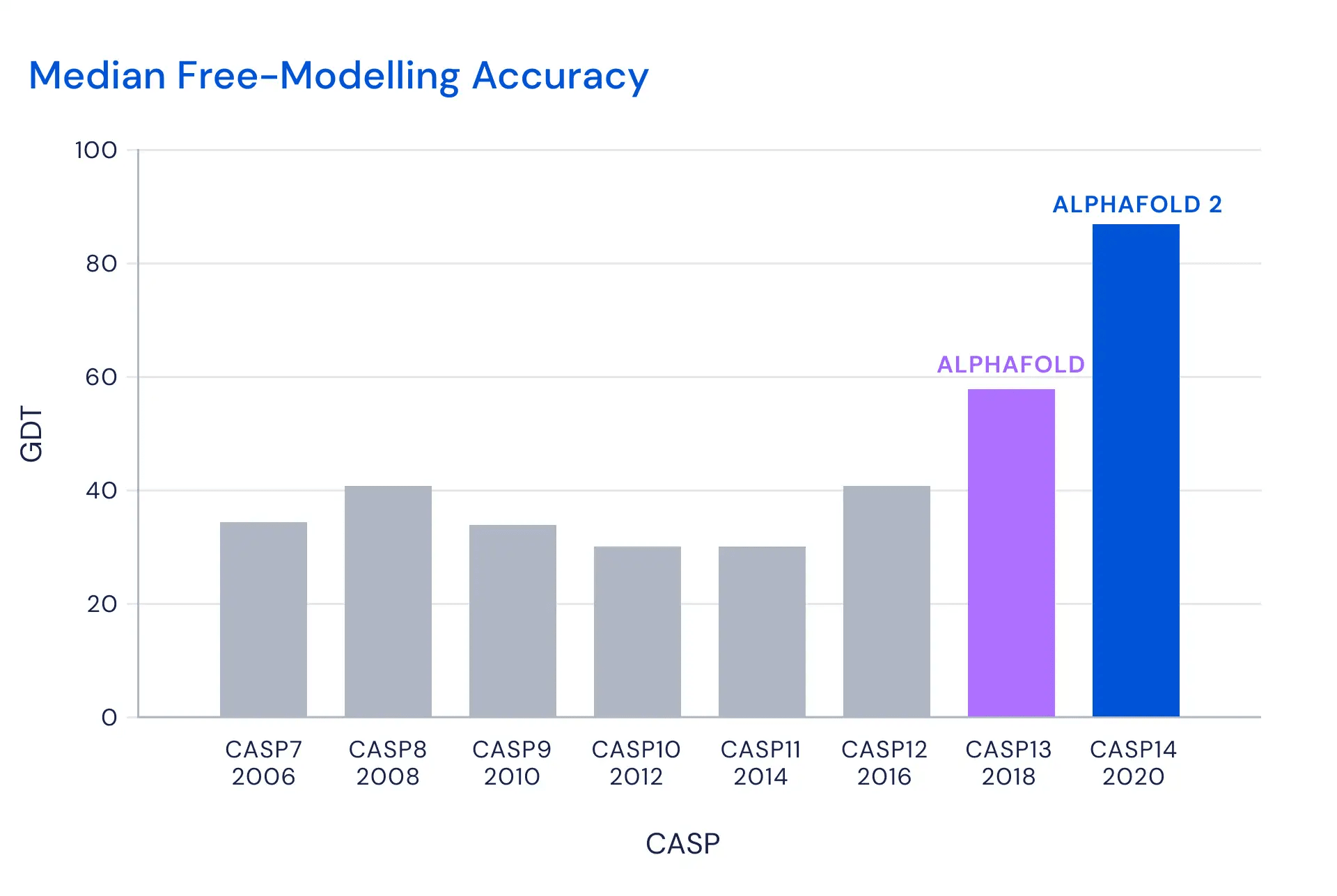
So how does AlphaFold work? It uses multiple deep neural nets to learn different functions relevant to each protein. One key function is a prediction of the final distances between pairs of amino acids. This guides the algorithm to the final structure. In one version of the algorithm (described in Nature and Proteins), they then derived a potential function from this prediction, and applied simple gradient descent—which worked remarkably well. (I can’t tell from what I’ve been able to read today if this is still what they’re doing.)
A general advantage of AlphaFold over some previous methods is that it doesn’t need to make assumptions about the structure. Some methods work by splitting the protein into regions, figuring out each region, then putting them back together. AlphaFold doesn’t need to do this.
DeepMind seems to be calling the protein folding problem solved, which strikes me as simplistic, but in any case this appears to be a major advance. Experts outside Google are calling it “fantastic”, “gamechanging”, etc.
Between protein folding and CRISPR, genetic engineering now has two very powerful new tools in its toolbox. Maybe the 2020s will be to biotech what the 1970s were to computing.
Congrats to the researchers at DeepMind on this breakthrough!
8 comments
Comments sorted by top scores.
comment by adamShimi · 2020-12-01T13:38:10.685Z · LW(p) · GW(p)
Thanks a lot for the explanation!
Notably, proteins embedded in the cell membrane—such as the ACE2 receptor that COVID-19 binds to—fold in the lipid bilayer of the cell and are difficult to crystallize.
Could you go into a bit more detail here? Why are these proteins so hard to measure? Can't you just "remove them from the membrane"? (Sorry if this is completely stupid, my knowledge of experimental biology is very limited)
Also, since you worked in the field, I'm curious about your take on the opinion that protein folding is not really useful, as articulated by John in my link post [LW(p) · GW(p)] of the DeepMind blog.
Replies from: Spiracular, jasoncrawford, None, JohnGreer↑ comment by Spiracular · 2020-12-01T17:57:39.315Z · LW(p) · GW(p)
To give a somewhat-simplified explanation...
TL;DR: In water, charged parts will tend to rotate outward. In neutrally charged hydrophobic environments, non-charged protein parts will try to face outward. Cell membrane is a partially-hydrophobic environment, so one of the standard protein-folding rules is inverted in its fatty-layer zone. When taken out of it, the protein may literally flip (and also clump, with both itself and other membrane proteins).
Trans-membrane (TM) proteins aren't woven, they're sewed (and then folded). They're threaded in and out of the membrane through a pore, as the ribosome prints them. (While the sewing is loose, not tight, sewing is almost exactly the right way to think about it.*)
Amino acids (AA) can be charged +, charged -, or roughly neutral depending on sequence/peptide. AAs are the component parts of the thread that folds into proteins, and a long string of +charged AAs can help make a whole region on that thread charged (or for 0charge, neutral).
Water molecules are charged and bi-polar. Near a strong charge, they'll rotate their faces like a magnet to put their + end near a -, or - end near a +. Alternatively phrased: charged molecules are usually hydrophilic and drawn to water molecules (more like water-molecules are drawn to them... but same difference), while large non-charged molecules (ex: the main-body of fats and oils) are hydrophobic by comparison and tend to clump among themselves (something weak Van der Waals forces something).
(You know how oil and water self-assort, and don't mix? It's a lot like that.)
The membrane is charged on the surfaces (both inner and outer surface)**, but has a fatty, hydrophobic, neutrally-charged environment in its middle.
This alters the preferred/stable protein structure for the AAs in the (fatty, hydrophobic) threading-zone. In water, charged parts will tend to rotate outward. In neutrally charged hydrophobic environments, non-charged protein parts will try to face outward. While the charged AAs will... wish they had anywhere else to be; usually gravitating harder towards any water they can find, or to each other.
So, one of the standard protein-folding rules is inverted in the fatty-layer zone. Basically.
If you took them out of the embedded fat layer, they would literally flip (to the extent to which that was physically allowed), or that section would clump with the uncharged middle-portions of itself or other TM proteins.***
And since a sizable fraction of TM proteins are cross-membrane pores (transporting a specific molecule from one side of the membrane to the other), getting that cross-membrane portion accurately matters a LOT if you're trying to understand function.
* For once-through proteins, there's even a specialized trans-membrane starter-peptide-sequence "needle" that can tell the cell it's a TM protein in the first place. It's the first thing to get threaded through, and gets cut off after its job is done. See: Signal Peptide.
** Membrane is a "lipid bi-layer" technically; it's like a "charged-end - fat & fat - charged-end" sandwich
*** Side-note: TM proteins float around in the membrane like rafts. It's pretty cool.
↑ comment by jasoncrawford · 2020-12-01T18:10:24.280Z · LW(p) · GW(p)
I'm not a comp bio expert, but the core of @johnswentworth's argument seems to be that “protein shape tells us very little about [protein reactions] without extensive additional simulation”, and “the simulation is expensive in much the same way as the folding problem itself.”
Both true as far as I understand, but that doesn't mean those problems are intractable, any more than protein folding itself was intractable.
So I think you can argue “this doesn't immediately lead to massive practical applications, there are more hard problems to solve”, but not “this isn't a big deal and doesn't really matter” in the long run.
Replies from: johnswentworth↑ comment by johnswentworth · 2020-12-01T22:33:10.874Z · LW(p) · GW(p)
I agree with this answer - it is still likely to be a useful component in a simulation pipeline in the long run, but it's probably not going to revolutionize things as a standalone tool in the short run.
↑ comment by [deleted] · 2020-12-01T17:06:14.055Z · LW(p) · GW(p)
When you remove membrane proteins from the membrane without great care, they tend to either aggregate into amorphous hydrophobic goo which is impossible to measure well, or take on a shape that is not the shape they ordinarily take in this new different context.
comment by DirectedEvolution (AllAmericanBreakfast) · 2020-12-01T18:00:24.235Z · LW(p) · GW(p)
This blog post is from the 2018 CASP13, not this year, but it still does a good job of digging in to some of the nitty-gritty issues in evaluating the advance this represents and the questions about its utility for practical research.
comment by [deleted] · 2020-12-01T07:33:00.250Z · LW(p) · GW(p)
I very much look forward to analysis of this model, since what it is 'seeing' probably tells you something about that which is conserved that is deeper than sequence over very long evolutionary time scales.