How Close We Are to a Complete List of Imprinted Genes
post by Morpheus · 2025-04-19T18:37:57.074Z · LW · GW · 1 commentsContents
TL;DR Epigenetics and imprinting Why we need to know all imprinting regions and genes How imprinted genes work and how we measure them Have we identified all imprinted genes? Fixing paternal imprinting might be easier than fixing maternal imprinting PCR for methylated DNA Thoughts on the role of RNA in imprinting Some useful datasets and resources if someone else wants to look further into imprinting and epigenetics in the future References None 1 comment
This post summarizes some of the research I have been doing for Bootstrap Bio AKA kman [LW · GW] and Genesmith [LW · GW]
TL;DR
- We currently don’t have a list of all imprinted genes that are important in human development, but further long-read sequencing in adults and in the human placenta is going to get us close to that goal.
- Measuring DNA methylation in a low input DNA context is really annoying, because usually we can’t amplify methylated DNA. I explore the only way that appears tractable to me to amplify methylated DNA by combining Phi29 and DNMT1. This could be useful for building an atlas of early embryo development and for epigenetic preimplantation embryo screening.
- Correct imprinting seems particularly achievable in Hulk sperm
Epigenetics and imprinting
This post assumes basic familiarity with epigenetics. The next paragraph is a 1-paragraph summary, but I recommend reading these two excellent posts on epigenetics. If you know what DNA methylation, DNMT1 and imprinting are, you can safely skip those posts. You might also just do fine by using the AI-generated glossary.
The short explanation is that DNA methylation consists of small chemical marks on DNA that change which proteins can bind well to it. Together with other chemical marks on histones, which are “spools” that DNA is rolled around, these marks are how a cell knows that it is a skin cell, rather than a neuron. Imprinting is a phenomenon where the two sets of chromosomes in early mammal embryos behave differently depending on if they were inherited from the father or the mother side. Imprinting appears to be mostly due to differences in DNA methylation and histone marks. DNA methylation and histone marks are maintained across cell replications through an array of specific enzymes that both add, maintain (DNMT enzymes in the case of methylation) and remove marks (Tet enzymes in the case of DNA methylation). Approximate maintenance of methylation is achieved by a balance between the actions of these enzymes in ways that aren’t fully understood yet.
Why we need to know all imprinting regions and genes
All strong methods of germline engineering require turning cultured cells that are not fully totipotent, back into totipotent or naive pluripotent stem cells[1] that an embryo can grow from. We might get to such a state through different mechanisms, including in vitro oogenesis, spermatogenesis or maybe we are able to find a cocktail that turns cells naive directly. While the Yamanaka factors make it possible to turn cells into a more stem-cell like state, they are not fully totipotent. One distinction that is often made here is between naive and primed iPSCs. Naive iPSCs share some characteristics, but the most important one is that they can specialize into cells from all three germ layers and are similar to cells in the blastocyst. Currently all methods to create naive human pluripotent stem cells are prone to loss of imprinting. If we properly imitate oogenesis and spermatogenesis in vitro, imprints are also erased. So in both cases it is important that we know of and understand all imprints in order to preserve or reestablish them.
Loss of imprinting tends to be really bad for embryos and have non-negligible effects. For example loss of paternal ZDBF2 imprinting is associated with a 20% lower weight in 2 week year old mice and this effect persists in weaker form throughout development![2] Confusingly though, ZDBF2 is found on chromosome 2 and presumably if ZDBF2 has such large effects it would have been seen as a pattern in UPD 2 cases? But perhaps UPD2 is too rare for the same doctor to have seen multiple patients to connect the dots? I think it’s also plausible that imprinting generally has larger effects in mice. In mice there seems to be stronger competition between paternal and maternal line due to being less monogamous than humans (One sign of this is that IGF2R is always imprinted in mice, but only polymorphically imprinted in humans).
Thus if we want to make healthy embryos, we need to make sure that ALL important imprints are correct in early embryos.[3] For this goal, it would be great if we had a list of all existing imprinted genes and their imprinting control regions, so we can make sure currently developed techniques to maintain imprinting don’t overlook any crucial ones. In the rest of this post, I will do a deep dive through existing papers, to explain how far we are in documenting all existing imprinted genes and what methods we might use to better screen for them.
How imprinted genes work and how we measure them
So there are about ~200 imprinted genes. At least in mice the pattern seems to be that there is a bimodal distribution between genes that are heavily biased in expression (close to 0 expression from 1 parent) and the regular genes which are expressed in roughly equal ratios. There are some genes that are only a little biased (think 52/48) and these tend to occur close to existing imprinting regions (Edwards et al., 2023). I think it is fair for the purposes of germline engineering to treat little biased genes as not imprinted.
In humans, it appears that all imprinted genes are ultimately regulated through some differentially methylated region (DMR). A DMR in this context is a region that is methylated in the chromosome inherited from one parent, but not the other. When there are differences in methylation between different cell types, those are also often referred to as DMRs, but those are not interesting to us in this post.
As you can see in the image below from wikipedia, methylation tends to
be low in early embryonic development and then increase
post-implantation. It is not shown in this image, but methylation tends
to increase more in the inner cell mass compared to trophoblasts which
stay more unmethylated. For this reason there are some differentially
methylated regions that are only transient in the embryo (tDMRs), but
that stick around in the placenta.

The majority of methylated regions only become differentially methylated after fertilization (somatic DMRs or sDMRs) caused by some nearby DMR that was inherited from the oocyte or sperm (germline DMRs or gDMRs). In mice, some sDMRs are established through the histone mark H3K27me3 set in the oocyte which then become methylated in preimplantation embryos. That methylation is then lost again in postimplantation embryos, but persists in the placenta. Since these types of DMRs are not following the “canonical” pattern of an imprint established through a methylated gDMR, they are called non-canonical imprints. It is possible that there are imprints in humans that are established through histones, as in the non-canonical imprints in mice. But if that’s the case it is probably a rare phenomenon, otherwise Daskeviciute et al. (2025) would have found one of these DMRs when they went explicitly looking for them.
We have identified some of the proteins that are involved in maintaining imprints. For example ZFP57 and ZNF445 appear necessary to maintain maternally methylated DMRs. CTCF and the histone mark H3K4me3 can help maintain DMRs unmethylated.
Have we identified all imprinted genes?
One natural question to ask if we are interested in germline engineering is if we have already identified all imprinted genes or if we at least know all imprinted genes that are crucial for development.
One of the first ways how imprinted genes were discovered is through patients with uniparental disomy, a rare genetic disorder where for a specific chromosome the patient has inherited two copies of a chromosome from the same parent, but none from the other (often as the result of a “rescued” trisomy). For more info, see my post on UPD.
Unfortunately, there isn’t one well maintained list for all known imprints. But combining the list from geneimprint.com with the list from Tucci et al. (2019) is pretty close to such a list. For learning about how we know specific genes are imprinted I would also recommend the Catalogue of Parent of Origin Effects over geneimprint.com, because it tends to cite more sources and includes more rational for why a gene was included or not. The genes found in these lists have been identified either through UPD disorders or methylation and RNA sequencing in both mice and humans.
Here’s how to find imprinted genes through RNA sequencing: Find RNA transcripts that are more often expressed from one chromosome rather than another. It is possible to identify if RNA fragments are from different chromosomes through SNPs that are different between maternal and paternal chromosome. If we have DNA data from the parents we can even identify if they are maternally or paternally imprinted as was done by Jadhav et al. (2019).
Some imprinted genes might be hard to track through RNA expression in humans, specifically for genes that are only imprinted in some specific kind of neuron or similar.
If the gene is also not imprinted in mice, one way how we could still identify such an imprint is through screening the entire genome for differentially methylated regions with high coverage epigenetic sequencing. If we do such screening for a lot of diverse cell types, I’d be relatively confident that we did not miss any imprinted genes.
Well known DMRs tend to be around one thousand pairs long and contain about 50 CpG sites and are about ~0-10% methylated on one chromosome and ~90% on the other (Monk et al., 2018). These are easy to identify through bisulfite sequencing by searching for long stretches that are about 50% methylated on average. Obviously there could be the problem that the important DMRs that we haven’t discovered are shorter or perhaps they are on average 70% methylated, because one chromosome isn’t fully unmethylated. There is really no way we can be sure how many such DMRs there are without drowning in false positives as long as we do not phase bisulfite reads by chromosome (like Zink et al. (2018) does).
Fortunately, long-read sequencing has recently become more economical and long-read sequencing allows us to both read methylation while also allowing us to phase DNA reads. We also have long-read DNA data that has higher coverage than any bisulfite sequencing that has ever been done to date. Using phased nanopore reads Akbari et al. (n.d.) was able to identify about 50 novel large differentially methylated regions. All of these novel regions seemed to be polymorphic and not differentially methylated in all individuals. Akbari et al. (n.d.) also identify 17 of these imprints to be conserved in mice and monkeys. So I don’t think it is off the table that these imprinted regions are essential early in development, in the brain or the placenta, but appear “polymorphic” in blood and other tissues. What is also suspicious is that Akbari et al. (n.d.) identify some imprints (like ZNF714) as somatic DMRs because they show less than 50% methylation in blastocysts. My best guess is that ZNF714 actually has a gDMR on its promoter that just appears to be a little less methylated in the blastocyst stage, but still managed to stay maintained.
Kindlova et al. (2023) uses phased parent of origin assigned nanopore reads to investigate imprinting in 8 human placentas. Kindlova et al. (2023) find two DMRs on genes not previously known to be imprinted, that they describe in more detail in their paper, but supplementary table 4 lists at least another ~90 such DMRs that weren’t identified in previous studies that show affinity for ZFP57. It’s possible that a lot of these are false positives, but some of them are probably real. Additional placenta samples will hopefully show which of these are genuine in the future.
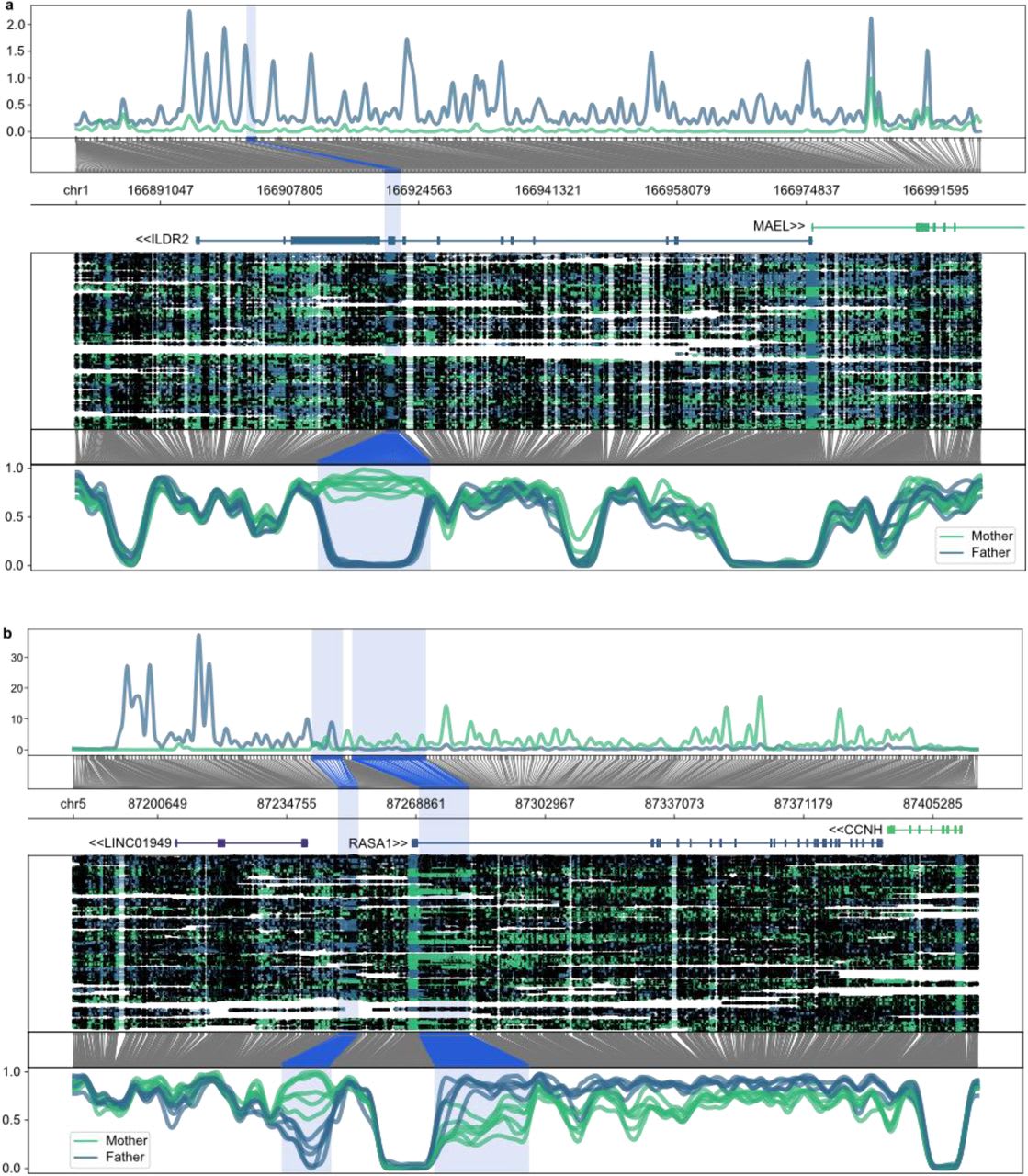
An image of the two novel DMRs discovered by Kindlova et al. (2023). The upper image shows the DMR on an exon of ILDR2 and the lower image shows the atypically looking DMR around the RASA1 promoter.
Fixing paternal imprinting might be easier than fixing maternal imprinting
Most imprints are maternally methylated and paternally unmethylated. This means creating Hulk sperm with correct imprinting might be comparatively easy, because we just erase all imprints by demethylating the cell on the way of turning it into a spermatogonial stem cell like cell and before transfering them into someone’s testicles, we only need to make sure that all imprints are properly demethylated and then perhaps using some more targeted strategy, like epigenetic Crispr to deal with the few remaining paternally methylated imprints.[4]
PCR for methylated DNA
Both for better understanding early embryos and for preimplantation screening of embryos, better methods of measuring methylation would be useful. Working with embryos means we are working with little DNA. 1 cell has about 6 picograms of DNA. Illumina sequencing methods need ~1000x more DNA than that to get adequate coverage and long-read sequencing methods have even higher input requirements.
The reason for this difficulty is that methylated DNA loses its methylation when amplified through common PCR methods. So traditionally the only way to read methylation is to chemically convert unmethylated cytosine into uracil (which both DNA polymerases and DNA sequencing methods read as thymidine) before amplifying DNA for downstream sequencing. The way this conversion has been done for the last 30 years is by treating DNA with sodium bisulfite. Not only does this degrade DNA (Tanaka & Okamoto, 2007), but it also reduces the complexity of the DNA sequence, making it harder to align reads. I have no lab experience and rudimentary chemistry knowledge, but a number often cited for how much DNA is lost through bisulfite treatment range from 70%-95% and there are some papers claiming to reduce the loss down to 25-35% under ideal conditions (Rajput et al., 2012; Yi et al., 2017). Recently, methods to convert the DNA via enzymes have also sprung up. Chatterton et al. (2023) try this in a single cell sequencing setting, but they end up losing more DNA through enzymatic conversion than through regular bisulfite sequencing. Overall both methods do not solve the problem that any procedure that we perform before amplifying the DNA is going to lead to unrecoverable losses in breadth of coverage.
So for this reason, if we want breadth of coverage with ultra-low input DNA, we need a method to amplify our methylated DNA. I see one tractable way to do this that hasn’t been extensively tried before: Use the same molecules human cells use, the enzyme DNMT1 and the reagent SAM to maintain methylation during PCR. For both of these there don’t exist alternative natural proteins that can withstand the high temperature during regular PCR, since methylated cytosine aminates too often at high temperatures, so bacteria that can withstand high temperatures use different chemical modifications to DNA. So we can only use polymerase that work well under low temperature (<40°C), which narrows down the DNA polymerase we can use to Phi29.[5] Liu et al. (2020) tried and patented[6] this idea. From their Figure3a and S10 I infer that DNMT1 seems to do de-novo methylation at a rate of 1-2% per replication and DNMT1 seems to fail to maintain methylation a similar fraction of the time.[7] That is a high false positive and false negative rate, but not fatally so if we get more coverage in return.[8] Overall their results are confusing in some areas, so I remain unsure if this method is any good. For example, Liu et al. (2020) get lower breadth coverage out of their amplified 10pg samples, compared to their unamplified controls.[9]
If it turns out the idea above isn’t really tractable or Liu et al. (2020) aren’t willing to license their idea at a reasonable price, my best guess is that methylation sequencing isn’t going to be high coverage enough to be of good use for pre-implantation embryo screening and (long or short-read) RNA sequencing might be more useful ¯\(ツ)/¯. Possibly the enzyme conversion method TAPS could still be worth it once commercially available, since it only converts methylated Cs to Uracil and thus keeps most of the sequence complexity intact. This blogpost also goes through some more intractable methylation sequencing strategies.
Thoughts on the role of RNA in imprinting
- Long non-coding RNA
- The textbook example for the role of RNA in epigenetics is the Xist gene, which silences one of the X chromosomes in humans and other mammals.
- Such long non-coding RNAs are sometimes used in imprinting regions to suppress multiple adjacent genes in cis. Prototypical examples are KCNQ1OT1 and Airn in mice (and presumably KCNQ1OT1 functions similarly in humans). In fact, Xist appears to be imprinted in mice, leading to mostly the paternal X-chromosome being inactivated in the placenta[10].
- I don’t know of any cases where the initial methylation vanishes, but the expression is still long-term imprinted. Seeing an example like that would be really interesting, but so far I haven’t seen one and it seems long-term imprinting is always maintained through methylation.
- People have been measuring these non-coding RNAs and there are not THAT many of them around. If one of them was acting like memory in some imprint that doesn’t have methylation involved similar to Xist, we would know that long non-coding RNA.
- microRNA
- There are examples of direct trans effects of an imprint on the other allele, where the paternal allele produces an antisense microRNA that is breaking down the maternal mRNA for that imprinted gene (Haig & Mainieri, 2020). There are presumably more examples like this. We should not expect imprints to only have cis effects.
Some useful datasets and resources if someone else wants to look further into imprinting and epigenetics in the future
I learned the hard way that epigenetics is too new a field to get a good understanding by reading the textbook, because being 13 years out of date is in fact making a big difference. For example for learning about histones and chromatin states, I would now recommend this YouTube video and after that just the wikipedia article.
When checking if a gene is a somatic or a germline differentially methylated region, it is important to have data about CpG methylation for both sperm and oocytes. Unfortunately oocytes are really expensive and even if studies end up using a handful of oocytes for bisulfite seq, the coverage per Oocyte is abysmal (1-10% of the genome are covered, for 42 oocytes, 12 oocytes or 9 oocytes (Hernandez Mora et al., 2023; Li et al., 2018; Zhu et al., 2018)). One exception is Okae et al. (2014), which used about 200 oocytes, but they didn’t provide me their processed data when I emailed them and their raw reads are in the Japan Genotype-Phenotype Archive, which is impossible to access if you don’t have an ethics board that has approved of whatever you want to do with that data. Fortunately, Akbari et al. (n.d.) used Okae et al.’s (2014) data to identify gDMRs in their paper and provided all the aggregated methylation data of those oocytes in the data repository associated with that study! That same data repository also contains a lot of other useful files for understanding imprinting, including both histone and methylation marks phased and separated by parent of origin. You can open all of these files in either igv, ucsc or the genome browser of your choice.
I also found some more phased methylation reads on the 1000 genomes project’s AWS bucket, that might be worth downloading.[11]
I can also recommend to read through some public peer review files, which gives a better impression what the state of the art is for academic outsiders like myself. The peer review of Akbari et al. (n.d.) and of some other studies is publicly available.
References
Akbari, V., Garant, J.-M., O’Neill, K., Pandoh, P., Moore, R., Marra, M. A., Hirst, M., & Jones, S. J. (n.d.). Genome-wide detection of imprinted differentially methylated regions using nanopore sequencing. eLife, 11, e77898. https://doi.org/10.7554/eLife.77898
Chatterton, Z., Lamichhane, P., Ahmadi Rastegar, D., Fitzpatrick, L., Lebhar, H., Marquis, C., Halliday, G., & Kwok, J. B. (2023). Single-cell DNA methylation sequencing by combinatorial indexing and enzymatic DNA methylation conversion. Cell & Bioscience, 13(1), 2. https://doi.org/10.1186/s13578-022-00938-9
Daskeviciute, D., Chappell-Maor, L., Sainty, B., Arnaud, P., Iglesias-Platas, I., Simon, C., Okae, H., Arima, T., Vassena, R., Lartey, J., & Monk, D. (2025). Non-canonical imprinting, manifesting as post-fertilization placenta-specific parent-of-origin dependent methylation, is not conserved in humans. Human Molecular Genetics, 34(7), 626–638. https://doi.org/10.1093/hmg/ddaf009
Edwards, C. A., Watkisnon, W. M., Telerman, S. B., Hulsmann, L. C., Hamilton, R. S., & Ferguson-Smith, A. C. (2023). Reassessment of weak parent-of-origin expression bias shows it rarely exists outside of known imprinted regions. In eLife. https://elifesciences.org/articles/83364; eLife Sciences Publications Limited. https://doi.org/10.7554/eLife.83364
Goyal, R., Reinhardt, R., & Jeltsch, A. (2006). Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Research, 34(4), 1182–1188. https://doi.org/10.1093/nar/gkl002
Greenberg, M. V. C., Glaser, J., Borsos, M., Marjou, F. E., Walter, M., Teissandier, A., & Bourc’his, D. (2017). Transient transcription in the early embryo sets an epigenetic state that programs postnatal growth. Nature Genetics, 49(1), 110–118. https://doi.org/10.1038/ng.3718
Haig, D., & Mainieri, A. (2020). The Evolution of <span class="nocase">Imprinted microRNAs</span> and Their RNA Targets. Genes, 11(9), 1038. https://doi.org/10.3390/genes11091038
He, C., ZHAO, B. S., NARKHEDE, P., Liu, C., & CUI, X. (2021). Method for highly sensitive DNA methylation analysis (Patent US11130991B2).
Hernandez Mora, J. R., Buhigas, C., Clark, S., Del Gallego Bonilla, R., Daskeviciute, D., Monteagudo-Sánchez, A., Poo-Llanillo, M. E., Medrano, J. V., Simón, C., Meseguer, M., Kelsey, G., & Monk, D. (2023). Single-cell multi-omic analysis profiles defective genome activation and epigenetic reprogramming associated with human pre-implantation embryo arrest. Cell Reports, 42(2), 112100. https://doi.org/10.1016/j.celrep.2023.112100
Jadhav, B., Monajemi, R., Gagalova, K. K., Ho, D., Draisma, H. H. M., van de Wiel, M. A., Franke, L., Heijmans, B. T., van Meurs, J., Jansen, R., Hoen, P. A. C. ‘t, Sharp, A. J., Kiełbasa, S. M., GoNL Consortium, & BIOS Consortium. (2019). RNA-Seq in 296 phased trios provides a high-resolution map of genomic imprinting. BMC Biology, 17(1), 50. https://doi.org/10.1186/s12915-019-0674-0
James. (2014). 5mC-PCR: Preserving methylation status during polymerase chain reaction. In Enseqlopedia.
Kindlova, M., Byrne, H., Kubler, J. M., Steane, S. E., Whyte, J. M., Borg, D. J., Clifton, V. L., & Ewing, A. D. (2023). An allele-resolved nanopore-guided tour of the human placental methylome (p. 2023.02.13.528289). bioRxiv. https://doi.org/10.1101/2023.02.13.528289
Laird-Offringa, I. A., Asong, J., Campan, M., Chen, P.-H., Marconett, C. N., & Haworth, I. S. (2016). Accurate in vitro copying of dna methylation (Patent US20160130643A1).
Li, L., Guo, F., Gao, Y., Ren, Y., Yuan, P., Yan, L., Li, R., Lian, Y., Li, J., Hu, B., Gao, J., Wen, L., Tang, F., & Qiao, J. (2018). Single-cell multi-omics sequencing of human early embryos. Nature Cell Biology, 20(7), 847–858. https://doi.org/10.1038/s41556-018-0123-2
Liu, C., Cui, X., Zhao, B. S., Narkhede, P., Gao, Y., Liu, J., Dou, X., Dai, Q., Zhang, L.-S., & He, C. (2020). DNA 5-Methylcytosine-Specific Amplification and Sequencing. Journal of the American Chemical Society, 142(10), 4539–4543. https://doi.org/10.1021/jacs.9b12707
Monk, D., Morales, den Dunnen, Russo, Court, Prawitt, Eggermann, Beygo, Buiting, Tümer, & and. (2018). Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics, 13(2), 117–121. https://doi.org/10.1080/15592294.2016.1264561
Okae, H., Chiba, H., Hiura, H., Hamada, H., Sato, A., Utsunomiya, T., Kikuchi, H., Yoshida, H., Tanaka, A., Suyama, M., & Arima, T. (2014). Genome-Wide Analysis of DNA Methylation Dynamics during Early Human Development. PLoS Genetics, 10(12), e1004868. https://doi.org/10.1371/journal.pgen.1004868
Rajput, S. K., Kumar, S., Dave, V. P., Rajput, A., Pandey, H. P., & Datta, T. K. (2012). An Improved Method of Bisulfite Treatment and Purification to Study Precise DNA Methylation from as Little as 10 pg DNA. Applied Biochemistry and Biotechnology, 168(4), 797–804. https://doi.org/10.1007/s12010-012-9820-7
Tanaka, K., & Okamoto, A. (2007). Degradation of DNA by bisulfite treatment. Bioorganic & Medicinal Chemistry Letters, 17(7), 1912–1915. https://doi.org/10.1016/j.bmcl.2007.01.040
Tucci, V., Isles, A. R., Kelsey, G., Ferguson-Smith, A. C., Tucci, V., Bartolomei, M. S., Benvenisty, N., Bourc’his, D., Charalambous, M., Dulac, C., Feil, R., Glaser, J., Huelsmann, L., John, R. M., McNamara, G. I., Moorwood, K., Muscatelli, F., Sasaki, H., Strassmann, B. I., … Ferguson-Smith, A. C. (2019). Genomic Imprinting and Physiological Processes in Mammals. Cell, 176(5), 952–965. https://doi.org/10.1016/j.cell.2019.01.043
Yi, S., Long, F., Cheng, J., & Huang, D. (2017). An optimized rapid bisulfite conversion method with high recovery of cell-free DNA. BMC Molecular Biology, 18(1), 24. https://doi.org/10.1186/s12867-017-0101-4
Zhu, P., Guo, H., Ren, Y., Hou, Y., Dong, J., Li, R., Lian, Y., Fan, X., Hu, B., Gao, Y., Wang, X., Wei, Y., Liu, P., Yan, J., Ren, X., Yuan, P., Yuan, Y., Yan, Z., Wen, L., … Tang, F. (2018). Single-cell DNA methylome sequencing of human preimplantation embryos. Nature Genetics, 50(1), 12–19. https://doi.org/10.1038/s41588-017-0007-6
Zink, F., Magnusdottir, D. N., Magnusson, O. T., Walker, N. J., Morris, T. J., Sigurdsson, A., Halldorsson, G. H., Gudjonsson, S. A., Melsted, P., Ingimundardottir, H., Kristmundsdottir, S., Alexandersson, K. F., Helgadottir, A., Gudmundsson, J., Rafnar, T., Jonsdottir, I., Holm, H., Eyjolfsson, G. I., Sigurdardottir, O., … Stefansson, K. (2018). Insights into imprinting from parent-of-origin phased methylomes and transcriptomes. Nature Genetics, 50(11), 1542–1552. https://doi.org/10.1038/s41588-018-0232-7
In which case we need a donor for the placenta. ↩︎
“At 2 weeks of postnatal age, male and female paternal-knockout mice were visibly smaller, with a 20% weight reduction (Fig. 6a,b,d,e) that affected all organs uniformly (Supplementary Fig. 8c). The unimodal distribution of the body weight data into 10% bin groups was consistent with high penetrance and low variance of the phenotype (Fig. 6f), but with a stronger effect in females (Fig. 6a–d). The undergrowth phenotype tended to minimize with age, but it was nevertheless persistent, as measured at 30 weeks (Fig. 6c).” (Greenberg et al., 2017) ↩︎
Unless we have really good reasons to believe that a particular imprint is not crucial for development. ↩︎
So far I know of only 2 paternal imprints where I am confident that they are not only established after fertilization and those are the IGF2/H19 DMR and the IG-DMR. There are probably more paternal imprints, that are important, but erased early on in development (possibly true of RASGRF1 in humans). ↩︎
Phi29 is already a common choice in Preimplantation embryonic testing and is used in commercial Kits like Repli-G from Quiagen. ↩︎
He et al. (2021) ↩︎
Although one can’t strictly separate replications with Phi29, since it is working continuously, but in the paper they claim to have amplified their 10pg sample by 100x. ↩︎
Because DNMT1 alone has a tendency to not only methylate semi-methylated, but also unmethylated cytosine (Goyal et al., 2006; James, 2014). We might also want to add UHRF1, which Laird-Offringa et al. (2016) claims significantly helps with this problem. They tried this in E. coli which do not have histones. If it works in E. coli my guess is that it should also be helpful during PCR. ↩︎
See table 2 in the paper’s appendix. Also for some reason their 100ng control ends up with lower genomic coverage than their 4ng control? Even though the 100ng control has a higher mapping ratio? ↩︎
I think this is why rodents turn off their X-chromosomes twice, but honestly I still don’t quite understand why this happens. ↩︎
You can find them on AWS by running
aws s3 ls s3://1000g-ont/ --recursive | grep ".bed" | grep -P "mat|pat"in the shell with AWS-cli installed. ↩︎
1 comments
Comments sorted by top scores.