What is epigenetics?
post by Metacelsus · 2022-11-06T01:24:05.350Z · LW · GW · 4 commentsThis is a link post for https://denovo.substack.com/p/what-is-epigenetics
Contents
What is genetics? What is epigenetics? Reading epigenetic marks Writing epigenetic marks Erasing epigenetic marks TL;DR: None 4 comments
Among all areas of biology related to my research, epigenetics is the one that is most commonly misunderstood, not only by the general public but even by other scientists. After being irritated one too many times,[1] I’ve decided to make a series of posts to explain what epigenetics really is, why it’s important, and how it’s misunderstood. I will also explain how epigenetics is important for my own research on making gametes from stem cells.
This first post covers the definition of epigenetics, and the basic biology of epigenetic marks.
What is genetics?
Before defining epigenetics, let’s start with a definition of genetics. Genetics is the study of genes, which are sequences of genetic material[2] that encode functional products.
Let’s take the IGF2 gene as an example.
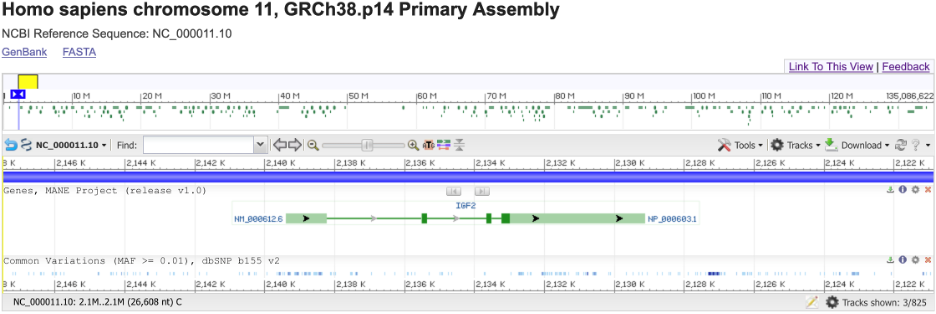
Depicted above is a region of human chromosome 11 containing the IGF2 gene, which encodes the IGF2 protein, an important growth factor for fetal development.[3] The boxes represent exons and lines represent introns. The darker green color is the protein-coding sequence, and non-coding (i.e. untranslated) regions are shown in lighter green. Arrows represent the direction of transcription.
The bottom of this image shows the location of common genetic variants (present at >1% frequency). If you look closely, you might notice that none of them are in the protein-coding sequence (the dark green boxes). This is not a coincidence, because nothing is ever a coincidence most mutations to essential proteins (including IGF2) are harmful and thus selected out of the population. However, there are several common mutations in non-coding regions of this gene.
To recap, genetics is the study of genes (such as IGF2) and the effects of genetic variation on their functions.
What is epigenetics?
Epigenetics is the study of epigenetic marks, which are changes to genetic material that alter gene expression, but do not change the genetic sequence. A decent analogy for epigenetic marks is CAPITALIZATION, bolding, or strikethroughs in text.
DNA methylation and histone modifications are the two kinds of epigenetic marks. Some people also consider long noncoding RNAs (such as those involved in X-chromosome inactivation) to be epigenetic marks. Although these RNAs are undoubtedly important for regulating gene expression, I would not classify them as epigenetic marks since they are not direct modifications to genetic material.
In vertebrate animals, the cytosine in CG sequences often has a methyl group attached, forming 5-methylcytosine. A CG sequence is also CG on the opposite strand, so the cytosines on both strands can be methylated.

To make things confusing, methylation at CG sequences is termed CpG methylation, the lowercase p standing for phosphate. 5-methylcytosine will pair with guanine just like normal cytosine, but it is not equivalent to cytosine in its interactions with DNA-binding proteins. Generally, CpG methylation suppresses the expression of nearby genes. CpG sites often cluster together to form “CpG islands” in important regulatory regions. Other organisms (invertebrates, plants, fungi, bacteria) have different ways of methylating DNA. I won’t get into them in this post series, but you should know that CpG methylation is not universal.
Modifications to histones are another important set of epigenetic marks. Histones are DNA packaging proteins, which form complexes called nucleosomes. DNA winds around nucleosomes sort of like thread around spools. The overall assembly of DNA and histones is known as chromatin.
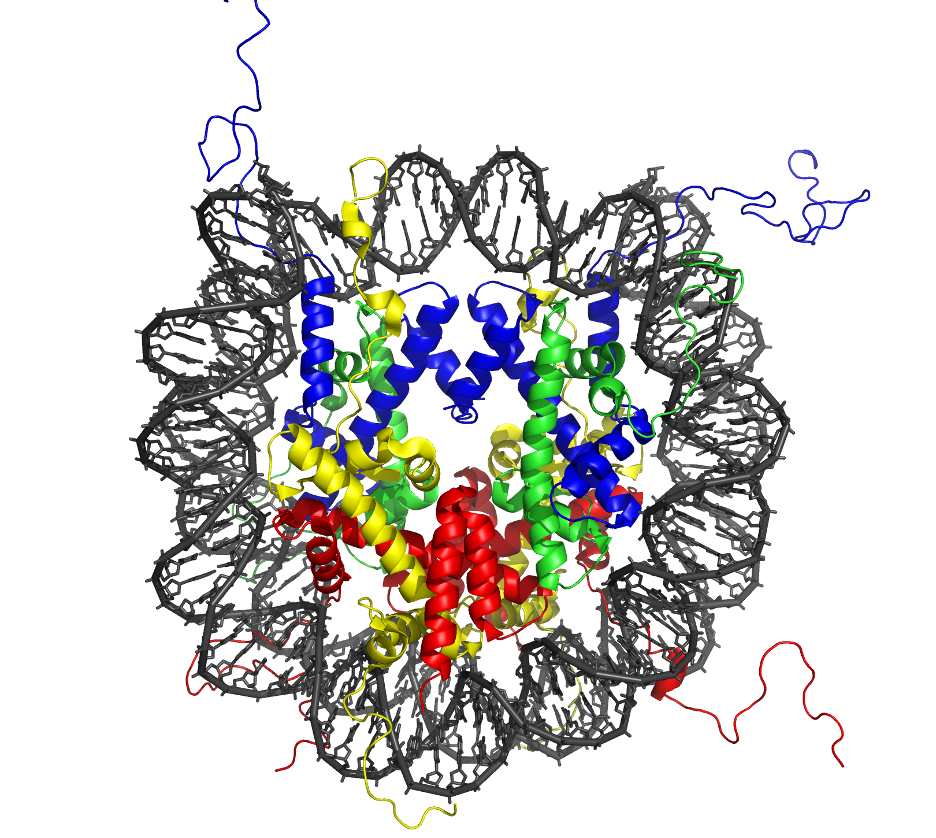
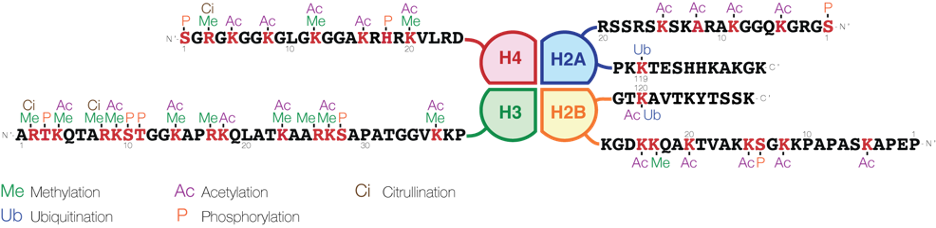
Chemical modifications to histones are important epigenetic marks that can have drastic changes on gene expression. For example, trimethylation of lysine 4 on histone H3 (known as H3K4me3) marks promoters of actively transcribed genes. However, methylation at other histone sites (such as H3K9 and H3K27) is repressive. Besides methylation, there is also a plethora of other histone modifications: acetylation, phosphorylation, ubiquitylation, sumoylation, crotonylation . . . the list goes on and on, and more are being discovered every year.
Let’s take another look at the IGF2 gene. Now I have added three additional display tracks related to epigenetic marks:
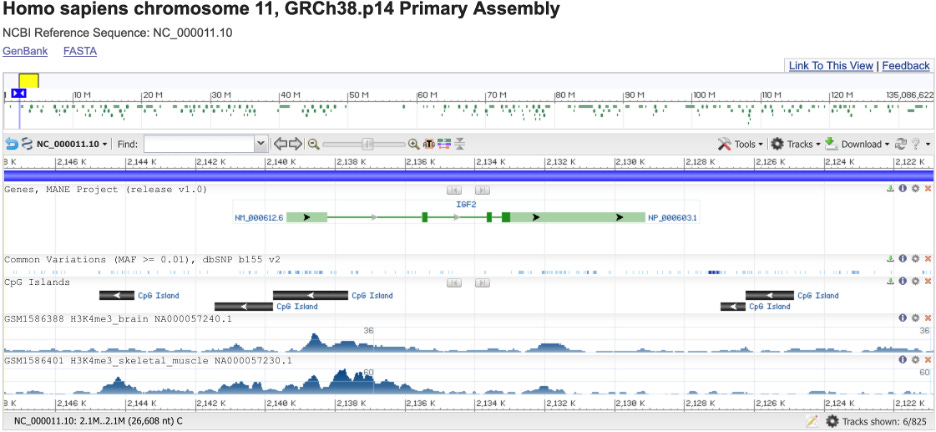
The “CpG Islands” track shows areas containing many CG sequences that could be methylated. Unfortunately, NCBI doesn’t have any information on the actual methylation status. The H3K4me3 tracks are more interesting. If you look closely, you may notice that the distribution of H3K4me3 is different in brain and skeletal muscle. This is not a coincidence: different types of cells have epigenetic marks in different places, and thus express different genes.
Reading epigenetic marks
Epigenetic marks are “read” by proteins that interact with DNA and/or histones. Many of these proteins have conserved domains that bind certain marks. A few of the many examples are:
- Methyl-CpG-binding domains bind methylated CpG sites
- Bromodomains bind acetylated histones
- Tudor domains bind methylated histones
These proteins are often transcription factors, which activate or repress gene expression. Furthermore, epigenetic marks also alter the physical properties of histones, particularly the electrostatic charge. Since DNA is negatively charged, marks that remove positive charges (e.g. acetylation) or add negative charges (e.g. phosphorylation) will make the histones bind to the DNA less strongly.
Scientists can also read epigenetic marks. For DNA methylation, the most common method is bisulfite sequencing, which chemically converts unmethylated cytosines to uracils, followed by sequencing of the DNA.[4]Any remaining cytosines observed in the sequence data must have been methylated.
Histone modifications are typically measured by chromatin immunoprecipitation sequencing (ChIP-seq), which uses an antibody to isolate histones bearing a particular epigenetic mark, and then sequences the associated DNA. Cut&RUN is a newer method that is conceptually similar but with higher sensitivity. These methods work well if and only if the antibody has strong on-target binding and low off-target binding. Various companies all claim that their proprietary antibodies are great, but it can often be challenging to find an antibody that actually works well, particularly for less commonly studied marks. Other methods such as ATAC-seq can measure whether DNA is loosely packaged (known as euchromatin, allowing for active transcription) or tightly packaged (known as heterochromatin, which represses transcription). This is closely related to measuring histone modifications, but not exactly the same.
Writing epigenetic marks
Epigenetic marks are written by specialized enzymes. Histone methylation and acetylation are established by methyltransferases and acetyltransferases. Each of these enzymes will typically be selective for only one particular target site. For example, H3K4 and H3K27 are methylated by different sets of enzymes. Histone phosphorylation is established by kinases. One important example is phosphorylation of serine 139 on H2AX, forming a modification known as γH2AX. This modification is added by the kinase ATM at sites of DNA damage, and subsequently recruits DNA repair enzymes.
DNA is methylated by DNA methyltransferases. In mammals, these are DNMT1, DNMT3A, and DNMT3B. DNMT3A and B are de novo methyltransferases, which means they can methylate fully unmethylated CpG sites. These enzymes are the ones responsible for determining where DNA methylation is added. Different proteins can recruit DNMT3 to sites that need to be methylated.

DNMT1 is a maintenance methyltransferase, which binds hemimethylated CpG sites and adds a methyl group onto the unmethylated cytosine. This is necessary to maintain DNA methylation after cell division, since newly synthesized DNA is always unmethylated. Most cells express DNMT1 and maintain methylation at the same sites over multiple rounds of cell division. However, the exceptions are extremely important, and I’ll discuss them in a later post.
Many epigenetic writer proteins recognize particular DNA sequence motifs. For example, promoter sequences of highly expressed genes contain sequence motifs that are bound by enzymes that deposit marks to activate gene expression. Also, certain DNA sequences known as insulators prevent epigenetic marks from spreading past them (as with PRC2, discussed below). These are just two of the many examples of genetics influencing epigenetics.
Notably, histone marks and DNA methylation also interact. Methylated DNA can recruit histone H3K9 methyltransferases which add additional repressive marks (H3K9me3). Likewise, DNMT3 enzymes contain an ADD domain that binds to unmethylated H3K4, meaning that the presence of H3K4me3 inhibits de novo methylation.
These marks all interact with themselves and each other by recruiting writer and eraser enzymes, forming complicated feedback loops. For example, the PRC2 complex methylates H3K27 to H3K27me3, and also binds to H3K27me3, which means that it spreads the methylation to adjacent areas of chromatin. However, it is stopped by H3K27ac because acetylated lysines cannot be methylated (and vice versa).
Researchers can write epigenetic marks at sites in the genome by attaching a writer enzyme onto a CRISPR protein such as dCas9.[5] The dCas9 attaches to a target DNA sequence and then the writer enzyme adds marks nearby. This can be useful in adding activating or repressive marks to turn target genes on or off.
Erasing epigenetic marks
Epigenetic marks are absent from newly copied DNA and newly synthesized histones, so in dividing cells they are lost by default unless actively re-written. This is very important, and we’ll come back to it in a later post.
There are also specialized proteins that can actively remove epigenetic marks. DNA methylation can be removed by TET enzymes which oxidize the methyl group to 5-carboxymethylcytosine. This is subsequently removed by thymine-DNA glycosylase[6] and the missing base is repaired using normal cytosine. Histone methylation can be removed by lysine-specific demethylases, and histone acetylation can be removed by histone deacetylases.
As with writing marks, researchers can also erase them at targeted sites by using eraser enzymes attached to dCas9.
TL;DR:
- Epigenetics is the study of epigenetic marks: modifications to genetic material that don’t affect the sequence, but control which genes get expressed.
- In mammals, these are DNA methylation and histone modifications.
- They can be read, written, and erased by specialized proteins.
- Newly copied DNA lacks epigenetic marks.
- Different cells have different patterns of marks and express different genes.
- Scientists can also read these marks through various sequencing-based technologies, and write or erase them using modified CRISPR proteins.
Next time: epigenetics of the mammalian germline, and how it explains why an egg can’t fertilize another egg and generate viable offspring.[7]
- ^
The latest example was this press release which took a finding in C. elegans and said "it may explain how a person's health and development could be influenced by the experiences of his or her parents and grandparents." WHICH IT DEFINITELY DOESN'T, C. elegans do things very differently from humans!
- ^
Typically DNA, but also sometimes RNA, for viruses with RNA genomes.
- ^
And also in several adult organs too. IGF2 is very interesting epigenetically and we’ll get into the details in the next post.
- ^
A related method, enzymatic methyl-seq, uses enzymes instead of bisulfite treatment.
- ^
d in dCas9 stands for “dead”. The amino acids that cut the DNA are mutated but it can still bind to the target.
- ^
This enzyme also removes the thymine from T:G mismatches. This is important because methylated cytosine can spontaneously deaminate and form thymine, and the cell needs a way of repairing this.
- ^
At least not without some hardcore bioengineering.
4 comments
Comments sorted by top scores.
comment by tgb · 2022-11-06T14:18:12.852Z · LW(p) · GW(p)
I work in a related field and found this a helpful overview that filled in some gaps of my knowledge that I probably should have known already and I’m looking forward to the follow ups. I do think that this would likely be a very hard read for a layman who wasn’t already pretty familiar with genetics and you might consider making an even more basic version of this. Lots of jargon is dropped without explanation, for example.
Replies from: Metacelsus↑ comment by Metacelsus · 2022-11-06T16:54:08.926Z · LW(p) · GW(p)
Yeah I probably fell victim to https://xkcd.com/2501/
Making a more simplified version is a good idea, and I'll probably do it after I'm done with the other posts.
Replies from: habryka4↑ comment by habryka (habryka4) · 2022-11-06T22:19:05.807Z · LW(p) · GW(p)
Yeah, as someone without a ton of pre-existing biology knowledge, there was definitely a lot of words and terms you used that I had to look up. I think I still got value out of it, but I do think my understanding of stuff is still pretty shaky given that I don't seem to have a lot of the foundations this post requires.
comment by trevor (TrevorWiesinger) · 2022-11-07T05:10:35.441Z · LW(p) · GW(p)
This information is extremely helpful to me. I can't say why because that is an infohazard (and one of the more serious ones), but rest assured that there is nothing at all wrong with posting this and more, and that I will make sure that it helps some extremely important people accomplish some extremely important things.