Model of psychosis, take 2
post by Steven Byrnes (steve2152) · 2023-08-17T19:11:17.386Z · LW · GW · 13 commentsContents
1. Introduction 2. Background: My “Model of psychosis, take 1” from earlier 3. Three areas where that first model seemed to be missing something important 4. My “Model of psychosis, take 2” 5. Nice things about this new model 5.1 Slow variations in the B/A ratio are at least a priori plausible, because B & A come from different neurons in different cortical layers (Layer 5 and Layer 2/3 respectively) 5.2 At least one paper seems to suggest that antipsychotics are better at suppressing layer 5 signals than layer 2/3 signals 6. Things I’m still unsure about 6.1 Which D2 receptors explain how antipsychotics work? 6.2 What about other causes of psychosis? None 13 comments
(I am not by any stretch of the imagination an expert on psychosis. This is more like “live-blogging my thinking as I go”. I’m hoping to spur discussion and get feedback and pointers.)
1. Introduction
I suggested a model of psychosis in my blog post “Schizophrenia as a deficiency in long-range cortex-to-cortex communication”, Section 4.2 [LW · GW] last February. But it had some problems. I finally got around to taking another look, and I think I found an easy way to fix those problems. So this post is the updated version.
For the tl;dr, you can skip the text and just look at the two diagrams below.
2. Background: My “Model of psychosis, take 1” from earlier
The following is what I was proposing in “Schizophrenia as a deficiency in long-range cortex-to-cortex communication”, Section 4.2 [LW · GW]:
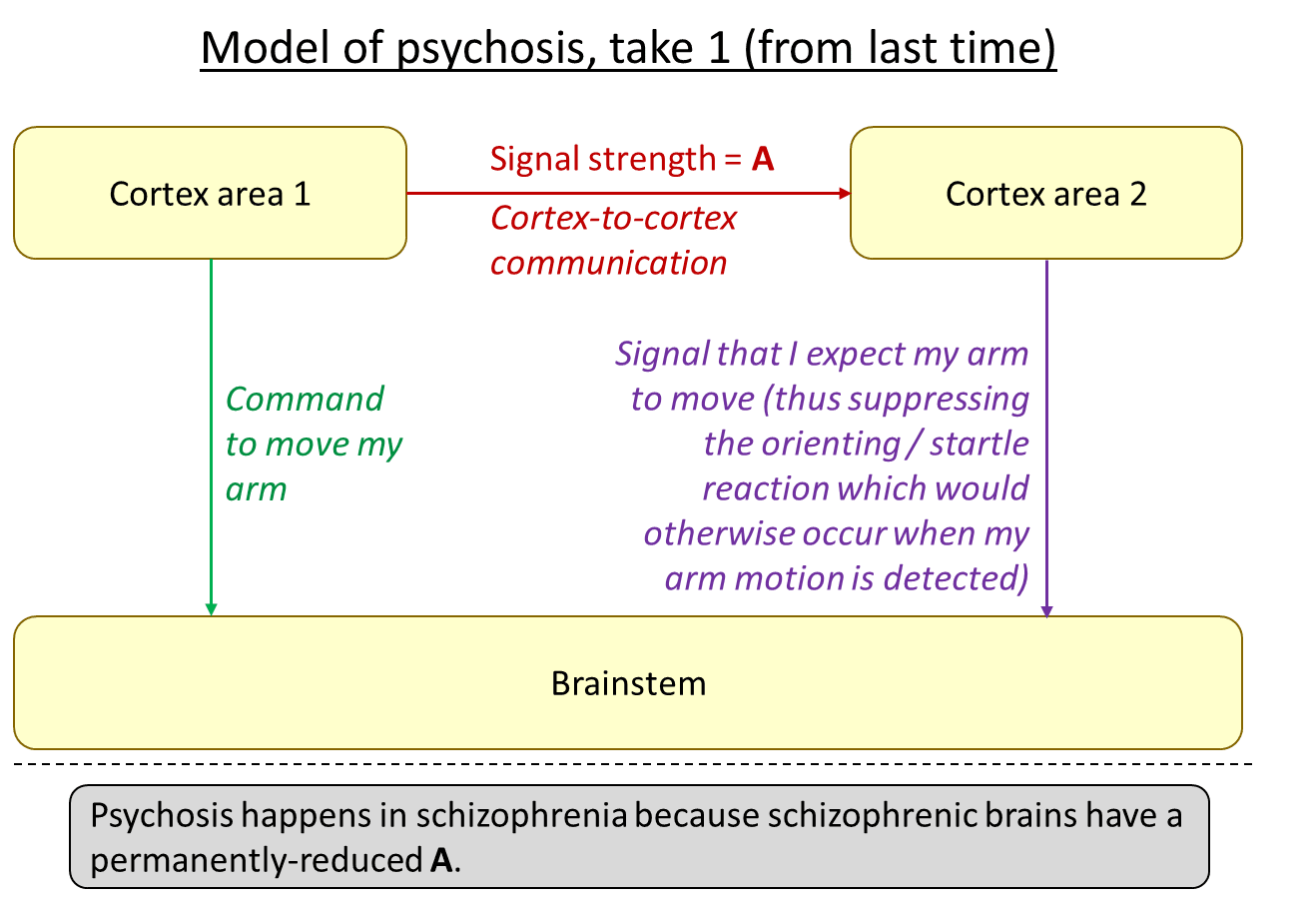
The idea is that, in psychosis, the green arrow is active and effective, but the red arrow isn’t, and therefore neither is the purple arrow. The result might be a kind of feeling that my arm was moved by an external force. That’s just one example—for different types of cortical outputs on the left, the corresponding first-person experience of psychosis would be different. But I claim that all symptoms of psychosis broadly fit into this template (update: I elaborated on “all symptoms of psychosis” in this comment [LW(p) · GW(p)] and also Section 7.5 of this later post [? · GW]).
3. Three areas where that first model seemed to be missing something important
- As I understand it, psychosis can come and go in people with schizophrenia, whereas (I believe) the deficiency in cortex-to-cortex communication is characteristic of schizophrenic brains, and is structural and permanent (absent future advances in medical technology).
- Antipsychotics reduce psychosis, but the diagram above offers no way to explain that.
- Psychosis can occur in other conditions besides schizophrenia. I think the most common example is the manic phase of bipolar. The diagram above cannot explain that.
4. My “Model of psychosis, take 2”
(The only change to the top part of the diagram is the new green text on the left that says “Signal strength = B”.)
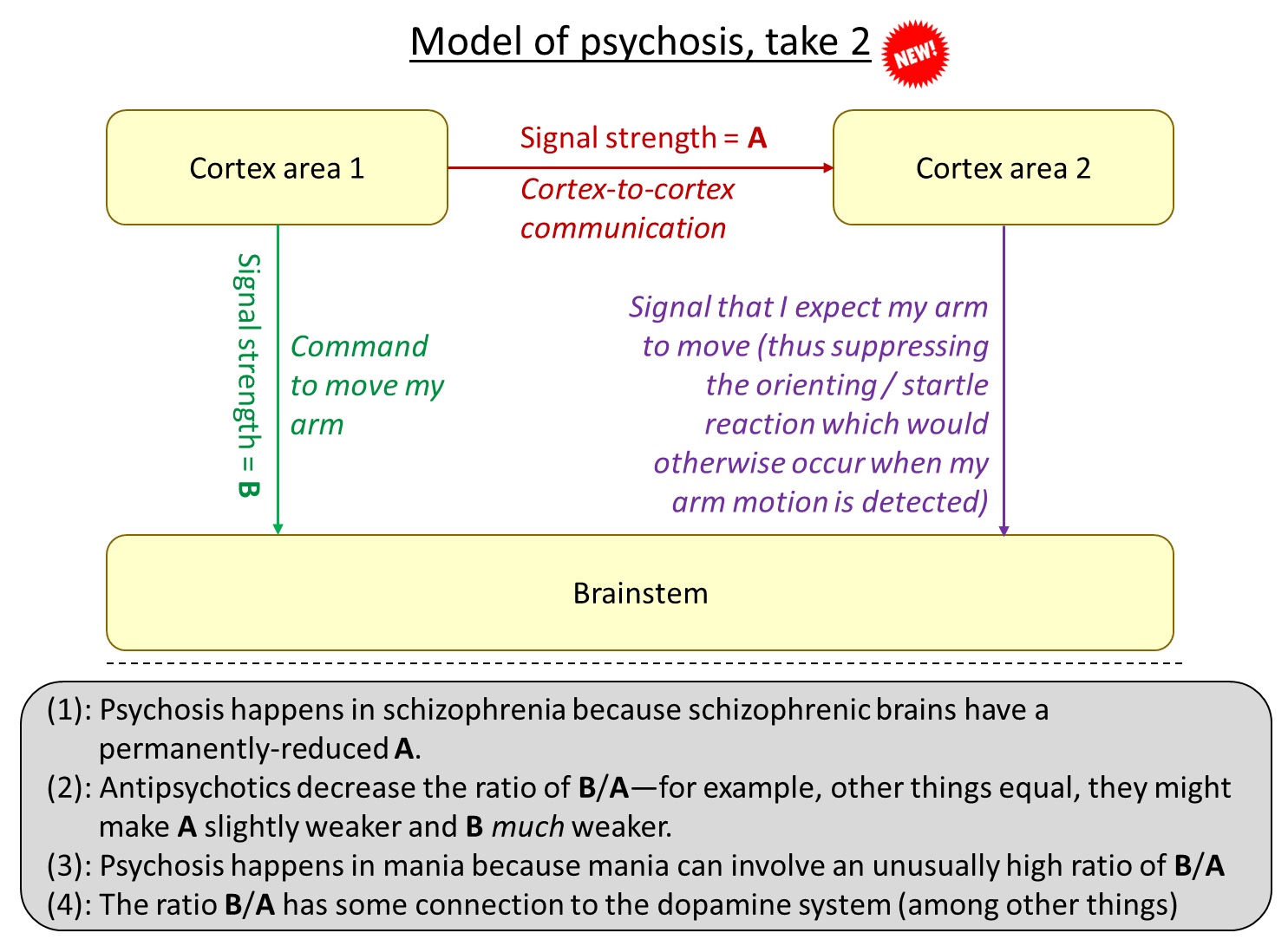
Think of a part of the cortex as having an adjustable “volume”, in terms of how strongly and clearly it is announcing what it’s up to right now. For example, if the thought crosses your mind that maybe you might move your finger, then you might find (if you look closely and/or use scientific equipment) that your finger twitches a bit, whereas if you strongly intend to move your finger, then your finger will actually move.

Anyway, imagine a big black analog slider for the “volume” of some part of cortex (“Cortex Area 1” in that diagram).
- Make a mark next to the slider at the volume threshold where Cortex Area 1 is “shouting” loud enough that its A messages start getting through and having an effect.
- Separately, Make a mark at the volume threshold where Cortex Area 1 is “shouting” loud enough that its B messages start getting through and having an effect.
…To avoid psychosis, we want the A mark to be at a lower volume setting than the B mark. That way, there will be no possible “volume level” in which the B messages are transmitting but the A messages are not.
(UPDATE: A bit more discussion in Section 7.5 of this later post [? · GW].)
5. Nice things about this new model
5.1 Slow variations in the B/A ratio are at least a priori plausible, because B & A come from different neurons in different cortical layers (Layer 5 and Layer 2/3 respectively)
I think it’s very plausible that the ratio B/A is a parameter that can vary, because:
- Signal B is sent exclusively by a subset of neurons in Layer 5 of the cortex
- Signal A is sent at least partly (and maybe mostly?) by a subset of neurons in Layer 2/3 of the cortex
- In general, different cortical layers have different types of neurons with different inputs, different relations to the dopamine system, etc.
So, a theory that involves long-term variation in the B/A ratio is at least plausible.
5.2 At least one paper seems to suggest that antipsychotics are better at suppressing layer 5 signals than layer 2/3 signals
See Heindorf & Keller 2023. They found that “clozapine…decreased correlations of cortical activity in [layer 2/3] excitatory neurons. However, this reduction was significantly weaker than the reduction we observed in … [layer 5 intratelencephalic] neurons”. (The p-values for this particular comparison were for short-range correlations and for long-range).
If I’m understanding the paper correctly (a big “if”!), this is suggestive and encouraging, but not clear support for my theory, because, first of all, that paper was measuring the wrong type of layer 5 pyramidal neurons compared to the ones sending Signal B, and second of all, the kind of correlation that the authors were measuring could be caused by the layer 5 neurons having weak signals in general (such that spatial correlations quickly fall below the noise floor) but there are other possible causes too (directly related to spatial correlations—I think that’s what the authors believe is going on).
6. Things I’m still unsure about
6.1 Which D2 receptors explain how antipsychotics work?
The thing that every antipsychotic has in common is blocking dopamine D2 receptors. So presumably that’s how they work. But there are D2 receptors in neurons all over the brain. Presumably, a subset of those neurons-with-D2-receptors are the secret to how antipsychotics work, and the rest are related only to side effects. Which is which?
When I try to flesh out my model, the most simple and elegant story that I’ve thought of so far involves D2 receptors in the cortex playing the starring role. In particular, different cortical layers have different densities of D2 receptors, and the sign seems to be correct for antipsychotics to decrease the B/A ratio, if I’m not mistaken.
But that’s funny because practically everyone else seems to think that D2 receptors in the striatum are how antipsychotics work, I think? I was trying to figure out why people seem to believe that, but couldn’t figure it out. All the evidence I could find for antipsychotics working via the striatum was pretty weak and indirect. Please comment (or email me) if you know anything about this.[1]
6.2 What about other causes of psychosis?
For various reasons, I have a sorta vague rule-of-thumb in my head that, other things equal, more dopamine tends to increase the B/A ratio (and hence, beyond some threshold, to cause psychosis). That seems to nicely explain the psychosis associated with mania (I loosely associate mania with “lotsa dopamine”, at least in certain channels and with various caveats), and the fact that psychosis is a side-effect of L-DOPA treatment for Parkinson’s.
I’m more confused about psychotic depression. (In bipolar, I understand that psychosis is much more common in mania than depression, but it can happen in depression.) I normally think of depression as the “opposite” of mania, and involving unusually little dopamine, again in certain channels and with various caveats. So I’m a bit confused that psychotic depression can happen at all. I dunno. The dopamine system is just one of many things that has an impact on the B/A ratio anyway. Or maybe the purple signal on the right side of the diagram above isn’t getting through? Or maybe all the things I just said about dopamine are too oversimplified? Or maybe it’s a different story entirely.
(UPDATE OCT 2024: I now think that psychotic depression is “a different story entirely”. I think the delusions are just a stronger version of emotional reasoning, and the occasional hallucinations are a generic consequence of bursts of strong emotion. See Section 7.6 of this later post [? · GW] for details.)
- ^
Update 2023-08-24: A friend sent me this paper (free version) which seems to support my tentative beliefs that both (1) it is currently conventional wisdom that antipsychotics work by preventing dopamine from inhibiting D2-receptor striatal neurons, and (2) this conventional wisdom is wrong. …If I’m understanding it correctly. Thanks for the tip, and keep ’em coming!
13 comments
Comments sorted by top scores.
comment by stormykat (sandypawbs) · 2024-11-13T21:23:55.288Z · LW(p) · GW(p)
This post is really interesting!
Do you have any thoughts on why then does psychosis typically suddenly 'kick in' in late adolescence / early adulthood? (and why trauma correlates with it and tends to act as that 'kickstarter'?)
Also any thoughts about delusions? Like how come schizophrenic people will occasionally not just believe in impossible things but very occasionally even random things like 'I am Jesus Christ' or 'I am Napoleon'?
Replies from: steve2152↑ comment by Steven Byrnes (steve2152) · 2024-11-13T22:10:03.004Z · LW(p) · GW(p)
Thanks!
Do you have any thoughts on why then does psychosis typically suddenly 'kick in' in late adolescence / early adulthood?
Yeah as I discussed in Schizophrenia as a deficiency in long-range cortex-to-cortex communication [LW · GW] Section 4.1, I blame synaptic pruning, which continues into your 20s.
and why trauma correlates with it and tends to act as that 'kickstarter'?
No idea. As for “kickstarter”, my first question is: is that actually true? It might be correlation not causation. It’s hard to figure that out experimentally. That said, I have some discussion of how strong emotions in general, and trauma in particular, can lead to hallucinations (e.g. hearing voices) and delusions via a quite different mechanism in [Intuitive self-models] 7. Hearing Voices, and Other Hallucinations [LW · GW]. I’ve been thinking of “psychosis via disjointed cognition” (schizophrenia & mania per this post) and “psychosis via strong emotions” (e.g. trauma, see that other post) as pretty different and unrelated, but I guess it’s maybe possible that there’s some synergy where their effects add up such that someone who is just under the threshold for schizophrenic delusions can get put over the top by strong emotions like trauma.
Also any thoughts about delusions? Like how come schizophrenic people will occasionally not just believe in impossible things but very occasionally even random things like 'I am Jesus Christ' or 'I am Napoleon'?
I talk about that a bit better in the other post:
In the diagram above, I used “command to move my arm” as an example. By default, when my brainstem notices my arm moving unexpectedly, it fires an orienting / startle reflex—imagine having your arm resting on an armrest, and the armrest suddenly starts moving. Now, when it’s my own motor cortex initiating the arm movement, then that shouldn’t be “unexpected”, and hence shouldn’t lead to a startle. However, if different parts of the cortex are sending output signals independently, each oblivious to what the other parts are doing, then a key prediction signal won’t get sent down into the brainstem, and thus the motion will in fact be “unexpected” from the brainstem’s perspective. The resulting suite of sensations, including the startle, will be pretty different from how self-generated motor actions feel, and so it will be conceptualized differently, perhaps as a “delusion of control”.
That’s just one example. The same idea works equally well if I replace “command to move my arm” with “command to do a certain inner speech act”, in which case the result is an auditory hallucination. Or it could be a “command to visually imagine something”, in which case the result is a visual hallucination. Or it could be some visceromotor signal that causes physiological arousal, perhaps leading to a delusion of reference, and so on.
So, I dunno, imagine that cortex area 1 is a visceromotor area saying “something profoundly important is happening right now!” for some random reason, and independently, cortex area 2 is saying “who am I?”, and independently, cortex area 3 is saying “Napoleon”. All three of these things are happening independently and unrelatedly. But because of cortex area 1, there’s strong physiological arousal that sweeps through the brain and locks in this configuration within the hippocampus as a strong memory that “feels true” going forward.
That’s probably not correct in full detail, but my guess is that it’s something kinda like that.
comment by Lorec · 2024-10-08T05:03:35.961Z · LW(p) · GW(p)
Doesn't this all rely on the idea that
Command to move my arm
and
Signal that I expect my arm to move (thus suppressing the orienting / startle reaction which would otherwise occur when my arm motion is detected)
are sent through separate [if parallel] channels/pathways? What substantiates this?
Replies from: steve2152↑ comment by Steven Byrnes (steve2152) · 2024-10-08T13:15:56.118Z · LW(p) · GW(p)
Yup! One is motor cortex, the other is somatosensory cortex. I’ll DM you some references and other reasons to believe me. :)
Replies from: Lorec↑ comment by Lorec · 2024-10-08T21:07:47.654Z · LW(p) · GW(p)
No, when I say "in parallel", I'm not talking about two signals originating from different regions of cortex. I'm talking about two signals originating from the same region of cortex, at the time the decision is made - one of which [your "B" above] carries the information "move your arm"[/"subvocalize this sentence"] and the other of which [the right downward-pointing arrow in your diagram above, which you haven't named, and which I'll call "C"] carries the information "don't perceive an external agency moving your arm"[/"don't perceive an external agency subvocalizing this sentence"].
AFAICT, schizophrenic auditory hallucinations in general don't pass through the brainstem. Neither do the other schizophrenic "positive symptoms" of delusional and disordered cognition. So in order to actually explain schizophrenic symptoms and the meliorating effect of antipsychotics, "B" and "C" themselves have to be instantiated without reference to the brainstem.
With respect to auditory hallucinations, "B" and "C" should both originate further down the frontal cortex, in the DLPFC, where there are no pyramidal neurons, and "C" should terminate in the auditory-perceptual regions of the temporal lobe, not the brainstem.
If you can't come up with a reason we should assume the strength of the "B" signal [modeled as jointly originating with the "C" signal] here is varying, but the strength of the "C" signal [modeled as sometimes terminating in the auditory-perceptual regions of the temporal lobe] is not, I don't see what weight your theory can bear except in the special case of motor symptoms - not auditory-hallucination or cognitive symptoms.
Replies from: steve2152↑ comment by Steven Byrnes (steve2152) · 2024-10-09T13:02:40.676Z · LW(p) · GW(p)
I’m confused …
I was saying that, in this particular illustrated case, B comes from motor cortex and C comes from somatosensory cortex. I can’t tell whether you are agreeing or disagreeing with that. In other words: You seem to prefer a model where B and C come from the same cortical area, right? But are you saying that I’m wrong even about the motor case that I used as my example in the diagrams, or are you setting aside the motor case and arguing about different cases like auditory hallucinations?
It's true that the bottom box is not necessarily always the brainstem specifically. That whole diagram is just illustrating the motor case as an example. Hmm, well, actually, it’s not fully general even for the motor case—the bottom box could also be the spinal cord in certain motor-related cases. If I were to draw the diagram for other non-motor-related phenomena, the bottom box might instead be the hypothalamus or other things—basically, anywhere that the cortical output neurons (“layer 5PT”) send signals to.
DLPFC, where there are no pyramidal neurons
I don’t think this is true; where’d you see that?
Replies from: Lorec↑ comment by Lorec · 2024-10-09T17:19:42.343Z · LW(p) · GW(p)
Thanks for being patient enough to go through and clarify your confusion!
About the pyramidal cells - I should have been more specific and said that prefrontal cortex [as opposed to primary motor cortex] - AFAIK does not have output pyramidal cells in Layer V. Those are Betz cells and basically only the primary motor cortex has them, although Wikipedia (on Betz cells and pyramidal cells) tells me the PFC has any neurons that qualify as "pyramidal neurons", too; it looks like their role in processing is markedly different from the giant pyramidal neurons found in the primary motor cortex's Layer 5.
Pyramidal neurons in the prefrontal cortex are implicated in cognitive ability. In mammals, the complexity of pyramidal cells increases from posterior to anterior brain regions. The degree of complexity of pyramidal neurons is likely linked to the cognitive capabilities of different anthropoid species. Pyramidal cells within the prefrontal cortex appear to be responsible for processing input from the primary auditory cortex, primary somatosensory cortex, and primary visual cortex, all of which process sensory modalities.[21] These cells might also play a critical role in complex object recognition within the visual processing areas of the cortex.[3] Relative to other species, the larger cell size and complexity of pyramidal neurons, along with certain patterns of cellular organization and function, correlates with the evolution of human cognition. [22]
This is technically compatible with "pyramidal neurons" playing a role in schizophrenic hallucinations, but it's not clear how there's any correspondence with the A vs B [vs C] ratio concept.
Maybe I slightly misunderstood your original theory, if you are not trying to say here, in general, that there is an "action-origination" signal that originates from one region of cortex, and in schizophrenics the major data packet ["B"] and the warning signal ["C"] experience different delays getting to the target.
If you are postulating a priori that the brain has a capability to begin the "B" and "C" signals in different regions of cortex at the same time, then how can you propose to explain schizophrenia based on intra-brain communication delay differentials at all? And if it's just signal strength, rather than signal speed, why bring "A" into it, why not just have "B" and "C"?
My own view is: Antipsychotics block dopamine receptors. I think you're right that they reduce a ratio that's something like your B/A ratio. But I can't draw a simple wiring diagram about it based on any few tracts. I would call it a Q/P ratio - a ratio of "quasi-volition", based on dopamine signaling originating with the frontal cortex and basal ganglia, and perception, originating in the back half of the cortex and not relying on dopamine.
Illustration of how the Q/P idea is compatible with antipsychotics reducing something like a B/A ratio in the motor case: Antipsychotics cause "extrapyramidal" symptoms, which are stereotyped motions of the extremities caused by neurons external to the pyramidal [corticospinal] tract. As I understand it, this is because one effect of blocking dopamine receptors in the frontal lobe is to inhibit the activity of Betz cells.
Replies from: steve2152↑ comment by Steven Byrnes (steve2152) · 2024-10-10T18:17:24.439Z · LW(p) · GW(p)
I don’t think time-delays are important at all here.
The part where I wrote “we gradually increase the “volume” of a part of cortex…” is trying to pedagogically explain something; I wasn’t literally saying that the “volume” actually increases “gradually” in the brain.
I have now rewritten that paragraph to avoid all mention of time (i.e. “first” and “second”). I also added a new diagram. Hope that’s clearer now, for you and anyone else who reads this going forward! Sorry for any confusion and thanks for feedback.
I still think you’re wrong about various aspects of pyramidal cells, but maybe we’ll have to agree to disagree on some of the issues here. :)
Replies from: Lorec↑ comment by Lorec · 2024-10-12T16:32:23.860Z · LW(p) · GW(p)
Thanks for clarifying. The point where I'm at now is, as I said in my previous comment,
Replies from: steve2152if it's just signal strength, rather than signal speed, why bring "A" [cortico-cortical connections] into it, why not just have "B" ["quasi-volitional" connections] and "C" ["perceptual" connections]?
↑ comment by Steven Byrnes (steve2152) · 2024-10-12T17:07:19.105Z · LW(p) · GW(p)
Hmm, this seems really obvious to me, so maybe we’re talking past each other somehow.
In this oversimplified picture, we’re supposing that somatosensory cortex is tasked with trying to figure out exactly where and how the arm will be moving in the immediate future.
How does the somatosensory cortex do that? Well, it takes whatever input data it has access to, and builds a (very-short-term) predictive model that leverages that data.
The input data is critical. In general, in life, you can’t make predictions if you have absolutely nothing to go on. If you ask me to predict who’s going to win the football match, but don’t tell me which teams are playing, then all I can do is guess randomly. If you tell me the teams, I can do a bit better; if you tell me their records so far this season, I can do even better; if you tell me which players are injured today, then I can do better yet; etc. More relevant input data enables better predictions.
Anyway, what’s the input data that the somatosensory cortex can use for its predictions? Well, visual data is helpful—maybe you see that your hand is heading towards a wall. Proprioceptive data is obviously helpful—for example, if the arm is already fully extended then you can be confident that it won’t extend further. And then there’s data from all sorts of random slow interoceptive fibers, like muscle strain sensors or whatever. All that data and more is good and helpful for the somatosensory cortex to issue accurate predictions of exactly where and how the arm will be moving in the immediate future.
…But those predictions are still going to be way off if the somatosensory cortex isn’t getting any data indicating what motor cortex is up to. Like, let’s say some part of motor cortex decides it’s a good time to move my arm, and then starts sending appropriate signals to motoneuron pools or whatever. But also assume there’s no data going from motor cortex to somatosensory cortex. Then obviously the somatosensory cortex would have no idea what the motor cortex is up to, and would issue very bad predictions regarding how my arm is about to move. Right?
Replies from: Lorec↑ comment by Lorec · 2024-10-13T18:23:28.252Z · LW(p) · GW(p)
I agree that the somatosensory cortex [in the case of arm movements, actually mostly the parietal cortex, but also somewhat the somatosensory] needs to be getting information from the motor cortex [actually mostly the DLPFC, but also somewhat the motor] about what to expect the arm to do!
This necessary predictive-processing "attenuate your sensory input!" feedback signal, could be framed as "A [C]", such that "weak A [C]" might start giving you hallucinations.
However, in order for the somatosensory cortex to notice a prediction error and start hallucinating, it has to be receiving a stronger [let's say "D"] signal, from the arm, signifying that the arm is moving, than the "weak A [C]" signal signifiying that we moved the arm.
I don't think your theory predicts this or accounts for this anyhow?
My "Q/P" theory does.
[ "B" in your theory maps to my "quasi-volition", ie anterior cortex, or top-down cortical infrastructure.
Every other letter in your theory - the "A", "C", and "D" - all map to my "perception", ie posterior cortex, or bottom-up cortical infrastructure. ]
comment by Carl Feynman (carl-feynman) · 2023-08-18T17:12:59.185Z · LW(p) · GW(p)
An interesting theory! But I am confused by why you think that antipsychotics can’t act simply by increasing the strength of A-type signals. Why do they also have to affect the B-type signals? if A signals are disproportionately inhibitory, that would account for why a notorious side-effect of antipsychotics is stiffness and a general reluctance to do stuff. Your claim that the relapsing-remitting nature of schizophrenia doesn’t fit your first theory seems excessive. Lots of nerve problems are relapsing-remitting (eg mutiple sclerosis). And changes to the the balance of the brain that take weeks are very common (eg antidepressant efficacy, or heroin addiction).
An under activity of long-range fibers could perhaps explain the many abnormalities of visual perception seen in schizophrenics. This is a field full of odd symptoms, with as far as I know no unifying theory.
It seems like you’re only trying to explain one symptom of psychosis: the delusion of an external locus of control. Could you extend your theory to explain other symptoms?
For example, how do you explain hallucination, where somebody has a sensory impression that they know isn’t real (at least with mild psychosis). It seems like hearing voices is an overactivity of the connection from the language area to consciousness, rather than an underactivity.
Another major symptom that might be explained by your theory is delusions of reference, where stimuli that should be treated as minor are instead freighted with importance. Like “What is the ad on that bus that just drove by trying to tell me? Could it have been sent by God?” It’s not obvious what the cortical areas that are involved in this are. But you’re lots more informed than me; what do you think?
Replies from: steve2152↑ comment by Steven Byrnes (steve2152) · 2023-08-18T18:14:53.701Z · LW(p) · GW(p)
Thanks for your helpful comment!
But I am confused by why you think that antipsychotics can’t act simply by increasing the strength of A-type signals.
I think schizophrenia is characterized by a deficiency in long-range cortex-to-cortex communication, and I suppose that this deficiency can have a variety of causes in principle. For example, it was found in lab experiments that faulty glutamate signaling can cause schizophrenia—hence “the glutamate hypothesis of schizophrenia” which has been floating around for many decades. But that hypothesis never led to any viable drugs. And I think that’s because faulty glutamate signaling is not the usual cause of the deficiency.
Instead, I think in practice the deficiency is almost always caused by the long-range fibers (and/or their terminal synapses) just not being there in the first place, or at least not in sufficient numbers. I mention some direct evidence for that in section 5 of my previous post [LW · GW].
So basically, I think that cortex-to-cortex communication is just going down a lousy communication channel (noisy / low-bandwidth / low-ability-to-impact-downstream-areas / whatever). And that that problem is basically unfixable in almost all schizophrenic people, absent miraculous future medical advances. And it’s not the kind of problem that I expect to come and go, I think. It's structural.
But that communication channel, lousy as it is, can still transmit the information if the source-cortex is “shouting” long enough and loud enough. So I think the only viable approach that treating psychosis is to reduce the ratio B/A.
if A signals are disproportionately inhibitory, that would account for why a notorious side-effect of antipsychotics is stiffness and a general reluctance to do stuff.
Not sure about stiffness but “general reluctance to do stuff” is totally what I would expect from “less B”. I don’t follow how you connect it to A.
An under activity of long-range fibers could perhaps explain the many abnormalities of visual perception seen in schizophrenics.
I think so! I have an example in my previous post [LW · GW].
It seems like you’re only trying to explain one symptom of psychosis
Yeah, moving my arm is just one of many types of cortical output.
Another is subvocalizing / inner speech. I don’t understand all the details, but AFAICT producing a subvocalization involves sending signals to brainstem motor areas, and “hearing” your own subvocalization “activates similar areas of the auditory cortex that are involved in listening” (wiki). So my working hypothesis is that “hearing voices” in psychosis involves inner speech feeling like it’s external. Same diagram. (Low confidence though—again, I’m kinda confused about the mechanics of inner speech / subvocalization.)
Yet another type of cortical output is “attentional”—one part of the cortex can exert (metacognitive) control over the information flows and activities of other parts of the cortex (via signals to various subcortical areas). If you visualize a pen, it activates the same neurons in your temporal lobe as if you see a similar pen. You can flip back and forth by (among other things) voluntary metacognitive control. Nevertheless, for a neurotypical person, you don’t get confused about whether the thing you’re attending to is real or imagined. But I think things can get very screwy when one part of the cortex is manipulating the levers on metacognition / attention-control, and other parts of the cortex are not getting advanced notice that this metacognition / attention-control is happening. So basically, I think a visual hallucination would happen when the metacognitive / attention-control parts of the cortex (prefrontal probably) set part of the visual system (temporal lobe) to imagination-mode rather than attend-to-what’s-in-front-of-you mode, but other parts of the cortex don’t get the memo that we’re in imagination-mode right now, and they interpret the (imagined) current contents of the visual system as a reflection of what’s directly in sight and coming up through V1/V2/etc.
(Or something like that.)