The Lens, Progerias and Polycausality
post by johnswentworth · 2020-03-08T17:53:30.924Z · LW · GW · 8 commentsContents
Progerias None 8 comments
Fun fact: the lens of a human eye consists mostly of fiber deposits which are never broken down - they do not turn over. Furthermore, new fiber layers are constantly added throughout life, so the lens thickens linearly by about 25 microns per year. Starting at around 3.5mm in infancy, it reaches 5.5mm in old age.
The main clinical result of this is the practically-universal need for glasses for close-up vision in people over 55 years old.
(Source: Physiological Basis of Aging and Geriatrics; the section on the eye is one of the most detailed in the book.)
Besides being a simple, self-contained gear in its own right, the growth of the lens is a clear, knock-down example of an independent root cause [LW · GW] of one symptom of aging. We know exactly what’s accumulating in a nonequilibrium fashion: the fibers of the lens. It’s wildly unlikely that the growth of the lens is a root cause for other symptoms of aging - like wrinkles [LW · GW], atherosclerosis, Alzheimer’s, cancer, muscle degeneration, etc. So, we have a clear case for polycausality - at least for one symptom of aging.
That said, there’s a fair bit of evidence that most symptoms of aging share a common root cause, or at least a common intermediate. Qualitatively, many/most symptoms of aging in a wide variety of tissues:
- Look similar at the cellular level - there’s a loss of homeostasis, with cells dying off faster than they’re replaced, high levels of misfolded protein aggregates (a.k.a. junk), and markers of chronic inflammation
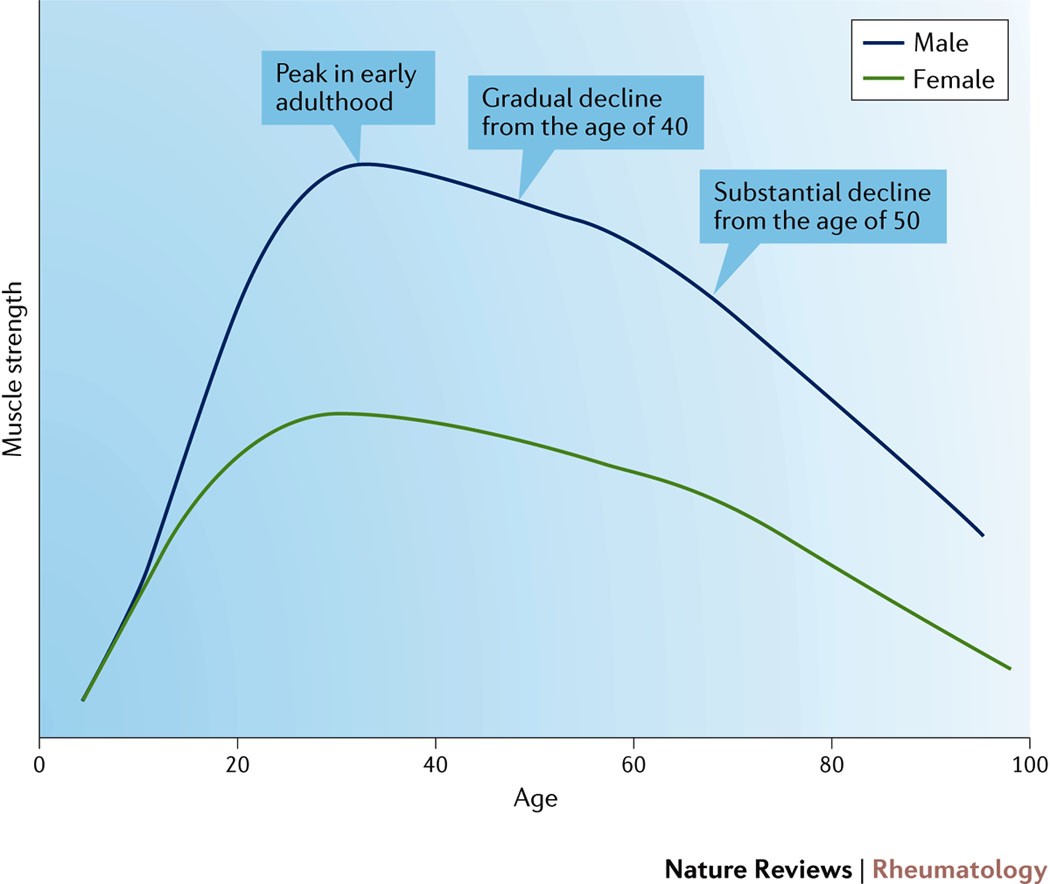
- Follow a similar population-level onset/progression timetable: no noticeable problems from youth through mid-twenties, gradual onset/progression throughout middle age, then rapidly accelerating breakdown around 50-60 years of age and older. Some examples: cancer incidence, muscle loss, atherosclerosis. Google a performance metric which declines with age, and you’ll probably see the pattern.
- Are correlated - someone who has one problem early is likely to have others early, and vice versa. See the literature on physiological/biological aging clocks for details.
{kind=link}
{kind=link}
{kind=link}
The growth of the lens does not follow this pattern - it’s just a straight-line linear growth starting from childhood, without any unusual role of chronic inflammation or misfolded proteins or other typical aging-associated characteristics. On the other hand, there are other contributing factors to old-age vision problems which do follow the usual pattern - for instance, the loss of pupil muscle mass.
Besides the growth of the lens, there are a handful of other possible root/intermediate causes of aging symptoms which don’t follow the usual pattern. None of them are as conclusive an example as the lens, but they may be involved in nastier diseases. In particular: the thymus is an organ which trains adaptive immune cells to distinguish pathogens from healthy host cells. That organ begins to shrink (called “thymic involution”) even in the first year of life, and steadily loses most of its mass by old age. I’ll likely have a full post on that later.
Progerias
One interesting source of evidence about common root causes of aging symptoms is accelerated aging diseases, a.k.a. progerias. I’ll talk about two: Werner Syndrome (WS) and Hutchinson-Gilford Progeria Syndrome (HGPS).
Werner syndrome is the progeria which most closely resembles true aging. People with WS develop normally through puberty, but then develop a laundry list of aging symptoms early:
- Gray hair
- Hair loss
- Wrinkles
- Skin hardening/tightening
- Loss of fat tissue
- Atrophy of gonads
- Cataracts
- Atherosclerosis
- Type 2 diabetes
- Muscle degeneration
- Bone loss
- Cancer
(you can find all this on the wikipedia page). Perhaps even more notable: changes in gene transcription associated with WS closely resemble the transcription changes associated with aging.
What causes this remarkably aging-like disease? Mutation of a gene called WRN (short for Werner), which is involved in repair of several types of DNA damage. The damage does still get repaired (otherwise people with WS wouldn’t be alive at all), but it’s slower, so presumably there’s a higher steady-state level of DNA damage. This is consistent with other lines of evidence which I may talk about in future posts: high levels of DNA damage are associated with aging.
The other type of progeria we’ll discuss is HGPS. HGPS also shows many aging-like symptoms:
- Hair loss
- Wrinkles
- Skin hardening/tightening
- Atherosclerosis
- Muscle degeneration
- Bone loss
But even more notable is the symptoms of aging which are not associated with HGPS, specifically:
- Cancer
- Arthritis
(Note: I didn’t comprehensively check every symptom of WS against HGPS, so don’t read too much into the differences between the two lists above.)
What would cause so many aging-like symptoms, but not cancer? HGPS is caused by mutation of a nuclear envelope protein; without it, the cell nucleus has a weird shape (striking picture here). The main result is that cells have trouble dividing - the folded-up nuclear envelope gets in the way of chromosome arrangement when the nucleus is supposed to divide. The mutation limits cell division, which we’d expect to lower homeostatic counts of a broad variety of cell types.
{kind=link}
Assuming that’s the main mechanism, we’d expect HGPS to show the symptoms of aging associated with cell loss - e.g. hair loss, muscle degeneration - but not the symptoms associated with biological stressors like DNA damage - e.g. cancer and inflammatory diseases like arthritis. For some symptoms which aren’t yet fully understood - e.g. wrinkles or atherosclerosis - HGPS is a hint that cell loss is probably a key mediating factor.
8 comments
Comments sorted by top scores.
comment by jimrandomh · 2020-03-08T19:10:42.351Z · LW(p) · GW(p)
This mostly makes sense to me; since DNA is at the root of many different regulatory mechanisms at once, cumulative DNA damage would be expected to gradually dysregulate everything. Since cell-count is one of the things being regulated, this predicts that DNA damage causes a superset of the symptoms of cell loss. And since DNA repair is itself one of the things being replicated, this would predict that DNA damage grows super-linearly.
(Interestingly, this implies that the safety screening done on food additives and other chemicals is more valuable than I previously thought; they look for DNA damage, on the theory that DNA damage leads to cancer.)
What's still confusing to me, though, is how little variance there is in the timeline of aging symptoms. This would seem to imply that there is something upstream of DNA damage and/or of cell loss, which does not behave like DNA-damage-causing-more-DNA-damage, but rather behaves more like lens fibers. I wonder what mechanism that might be?
(Also, let me just say that I really appreciate this sequence. While there is a fair amount of down-in-the-details biology work on aging, high-level conceptual explanations like this have been in short supply. I expect this sequence to meaningfully impact the number of people who wind up doing in-the-details work, and the quality of the targeting of that work.)
Replies from: johnswentworth↑ comment by johnswentworth · 2020-03-08T22:43:01.732Z · LW(p) · GW(p)
So, here's the surprising bit, which I think even a lot of biologists haven't fully absorbed: it's not DNA damage itself that's accumulating in a non-equilibrium manner. DNA damage events happen at a very fast rate, like thousands of times per day per cell (at least for that particular type of damage; there's several types). It's also repaired at a fast rate (true of all types, as far as I've seen). With that sort of half-life, if DNA damage were out of equilibrium, it would be out of equilibrium on a timescale much faster than aging. If DNA damage increases with age (and there's a lot of indirect evidence that it does), then it's a steady-state level that's increasing, which means that either the damage rate is increasing or the repair rate is decreasing.
In other words, DNA damage itself isn't the root cause - there is indeed something else upstream. You're exactly right on that. (Though, with respect to variance in timeline, bear in mind that many processes play out in parallel across cells - if cells "go bad" one by one, then large number statistics will smooth out the noise a lot.)
The most promising potential culprit I've heard about is transposons: "parasitic" DNA sequences which copy themselves and reinsert themselves into the genome. The human genome has loads of these things, or pieces of them - I've heard a majority of human DNA consists of dead transposons, though I don't have a reference on hand. Normally, they're actively suppressed. But the transposon theory of aging says that, every once in a while, one of them successfully copies. Usually it will copy into non-coding DNA, and then be suppressed, so there's no noticeable effect. But over time, the transposon count increases, the suppressor count doesn't increase, and eventually the transposons get out of control. The DNA damage is a side-effect of active transposons - one of the main ingredients of a transposon is a protein which snips the DNA, allowing the transposon itself to sneak in. In particular, this may be an issue in stem cells - most cells would enter apoptosis/senescence once DNA damage level gets high, but if a stem cell has a transposon count slightly below the cutoff, then it will produce cells which rapidly apoptose/senesce.
Anyway, I'm sure this sequence will get around to theories of the root cause of aging eventually. There's a number of them, although most have been ruled out.
Replies from: jimrandomh, alex-k-chen↑ comment by jimrandomh · 2020-03-08T22:52:02.445Z · LW(p) · GW(p)
I hadn't heard the transposon theory of aging before. If true, that would explain why aging hasn't been selected out by evolution: the transposons themselves have evolved, under different incentives than their host genome's incentives.
Replies from: johnswentworth, philh↑ comment by johnswentworth · 2020-03-08T23:30:15.661Z · LW(p) · GW(p)
Hold that thought - there's a post on evolution of aging coming up pretty soon. It's one of the better-understood areas, since we can get a ton of information by comparing across species.
↑ comment by philh · 2020-03-09T15:50:08.180Z · LW(p) · GW(p)
https://apomorphic.com/2020/01/12/why-we-age-2-nonadaptive presents a theory on that. Tldr (mostly from memory) would be that with no biological aging, we still have more kids when we're young, so evolution cares more about us when we're young, so there's very little selection pressure against mutations that only damage us when we're old, or especially which help us when we're young and harm us when we're old.
Replies from: willbradshaw↑ comment by willbradshaw · 2020-03-10T13:12:13.607Z · LW(p) · GW(p)
Thanks Phil. I should probably just put these on LessWrong to be honest.
The lens-growth phenomenon sounds like it might be a neat case of antagonistic pleiotropy as applied to developmental rates: a process calibrated to give good results in early adulthood might be selected for even if it gets wildly out of whack in later life. IIRC Williams gives the example of male Fiddler crabs, whose major claw grows faster than the rest of the body: the difference is calibrated to give them big sexy (but still manageable) claws in early adulthood but can severely impede movement in late life (I have not independently validated this example). One could imagine something similar happening here.
↑ comment by Alex K. Chen (parrot) (alex-k-chen) · 2020-10-25T18:04:27.025Z · LW(p) · GW(p)
>Usually it will copy into non-coding DNA, and then be suppressed, so there's no noticeable effect. But over time, the transposon count increases, the suppressor count doesn't increase, and eventually the transposons get out of control.
Wouldn't it expand the size of the genome and potentially affect the distance between promoters/enhancers and target genes, causing a loss in a cell's ability to appropriately regulate translation in response to perturbation?
I know some people (like genesis lung) who actively take lysine or antiretrovirals to suppress transposon activity - antiretrovirals may be aassociated with longevity./
Replies from: johnswentworth↑ comment by johnswentworth · 2020-10-25T18:21:54.701Z · LW(p) · GW(p)
Wouldn't it expand the size of the genome and potentially affect the distance between promoters/enhancers and target genes, causing a loss in a cell's ability to appropriately regulate translation in response to perturbation?
To some extent, though presumably the vast majority of copies will be into non-functional sequence, and copies into functional sequence will often result in a defective cell which is quickly removed. The expansion of the genome size shouldn't be significant until the count is already way out of control; a transposon is tiny compared to the whole genome.