Methods for strong human germline engineering
post by TsviBT · 2025-03-03T08:13:49.414Z · LW · GW · 28 commentsContents
Introduction Takeaways You would be surprised at how soon strong germline engineering can be made technically feasible. Disclaimers Terms Non-standard terms Standard terms Reproductive genomic vectoring Genomic vectoring (GV) Strong GV and why it matters Strength matters for reproductive genomic vectoring: Polygenic scores Editing and selection Other types of genomic vectoring (GV) Comparing editing and selection Editing produces out-of-distribution DNA Editing is already minimally working technology Headwind of mutation Fixing de novo mutations Bypassing natural integrity checks Selection makes similar children Selection and editing benefit differently from stronger PGSes Selection distributes Euclideanly, editing distributes Hammingly Selection beats editing, then editing beats selection Editing can reach more places than selection The ceiling of safe vectoring Many GV methods synergize Reproductive GV and epigenomic correctness (EC) The epigenomic correctness problem There should be more funding for epigenomic sequencing of germline cells: The near-miss hazard of epigenomic correction Going from 0.01 to 0.99 Biology's *scopes problem Methods to handle epigenomic correctness Takeaways: In vitro gametogenesis (IVG) Takeaways: The basic elements of gametogenesis IVG is a higher bar than epigenomic correction Other general remarks about IVG The general state of IVG research In vitro oogenesis (IVO) Takeaways: In vitro spermatogenesis (IVS) Takeaways: Using natural epigenomic correction No clear boundary between natural and artificial EC-making The timing problem with natural EC-making Alternative gonadal tissue Xeno gonads for full EC-making. Long-term culture of fetal gonads. Using natural reproductive DNA Natural EC interrupt methods Imprint maintenance problems Epigenetic CRISPR editing Donor embryo Hybrid natural and artificial EC-making How GV and EC interact A simple-ish and wrong model of combining GV and EC GV and EC are not conceptually separable Features of GV and EC methods that affect compatibility How feasible are different epigenomic correctness methods? How epigenomically disruptive are different GV methods? No epigenomic disruption Little epigenomic disruption Significant epigenomic disruption Haploid vs. diploid Summary of genomic vectoring methods Method: Simple embryo selection The power of simple embryo selection Super-duper-ovulation Method: Gamete selection Single gamete selection Gamete selection is mostly hypothetical But sperm selection might be doable The power of single gamete selection Double gamete selection The power of double gamete selection The limits of extreme gamete selection Method: Chromosome selection Implementation of chromosome selection Mechanical manipulation Whole cell fusion The power of simple chromosome selection Selecting a haploid genome Selecting a diploid genome Selecting an epigenomically correct haploid Selecting average chromosomes Selecting a diploid source of haploids The power of recombinant chromosome selection One-donor recombinant chromosome selection Many-donor recombinant chromosome selection Double recombinant chromosome selection With one mother, one father, and recombinant chromosome selection, you get more than +40 IQ points. Fractional haploid donation Method: Iterated recombinant selection Iterated embryo selection Iterated meiotic selection Auxiliary: Enhancements to meiotic methods Method: Iterated multiplex CRISPR editing DNA damage from editing Naive ESC editing Hulk sperm Magic rainbow sperm Conclusion Appendix: In vitro spermatogenesis studies Appendix: Cheap DNA segment sensing The problem of cheap DNA sensing Basic chromosome sensing Sensing crossovers An obstacle with sperm Cheap sensing at scale with FACS Sensing chromosome index Appendix: Best crossover What does recombination do to DNA? Math analysis of crossovers Simulation results of crossovers Refinements to the crossover model Non-uniform crossover points Multiple crossovers Appendix: The costs of iterated meiotic selection The power of segmented selection Breakdown at high granularity Segmented selection with one or more donors The cost of poor man's chromosome selection The cost of single-chromosome IMS Appendix: Detailed estimation of embryo selection Simple embryo selection simulations Analysis of simple embryo selection What is the distribution of embryos from one couple? What is the maximum scoring embryo from one couple? Appendix: Variation in chromosome length The issue with chromosome lengths Do chromosome lengths matter much? Why don't chromosome lengths matter much? Ignoring the 23rd chromosome Acknowledgements License None 28 comments
PDF version. Image sizes best at berkeleygenomics.org with a wide monitor. Twitter thread
Introduction
This article summarizes the technical pathways to make healthy humans with significantly modified genomes. These are the pathways that I'm aware of and that seem plausibly feasible in the next two decades. A short summary, in a diagram:
Annotated table of contents:
-
Reproductive genomic vectoring explains the general idea of human germline genomic engineering, and distinguishes editing and selection.
-
Comparing editing and selection talks about general differences between the two kinds of genomic vectoring methods.
-
Reproductive GV and epigenomic correctness (EC), Methods to handle epigenomic correctness, and How GV and EC interact discuss the epigenomic correctness problem in germline engineering—what it is, why it matters, and how to address it.
-
Summary of genomic vectoring methods gives an annotated table of contents for the following Methods sections. The Methods sections—on Simple embryo selection, Gamete selection, Chromosome selection, Iterated recombinant selection, and Iterated multiplex CRISPR editing—give more detail about each genomic vectoring method: what it is, obstacles, variations, and how powerful it is.
-
The appendices give additional technical information, if you're looking around and saying "I'm not in the weeds enough, I want to be more in the weeds.".
Here's a sneak peek about the strength of different genomic vectoring methods:
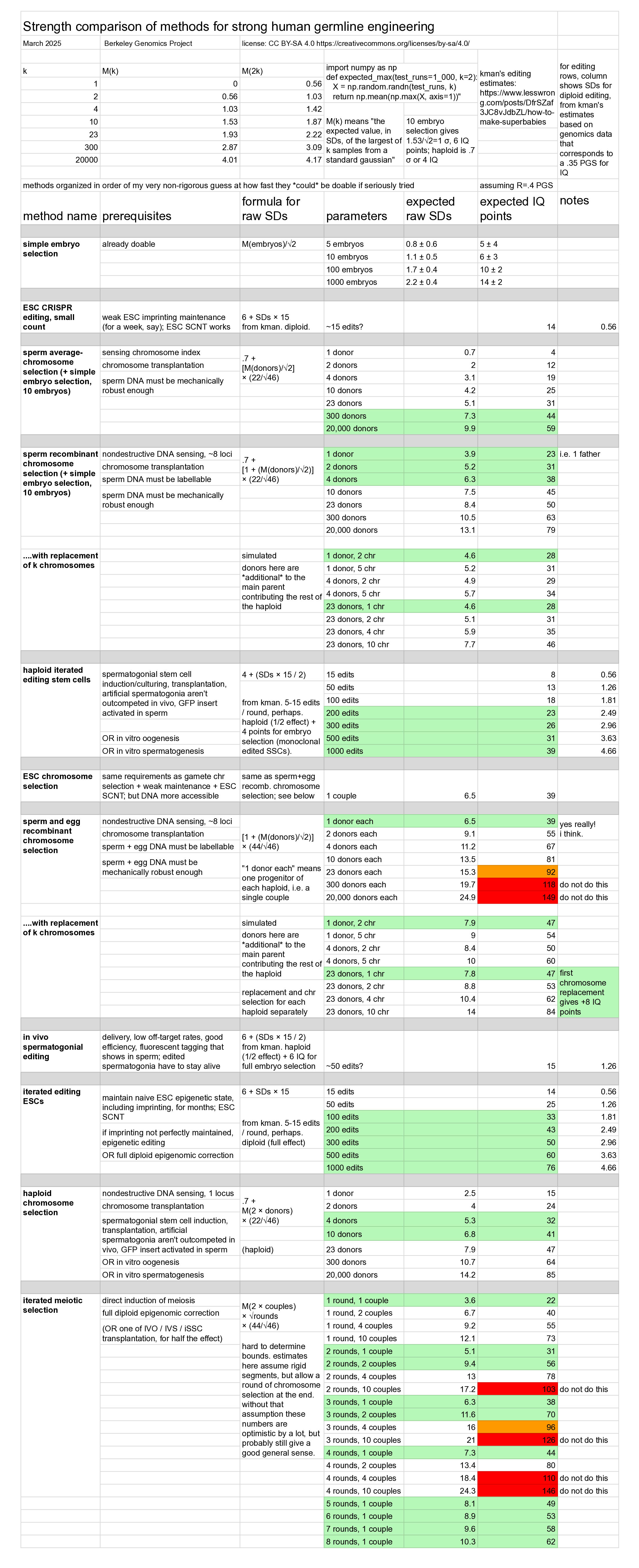
The list of specific methods that this table summarizes starts after the section Summary of genomic vectoring methods. This article is roughly organized from general to specific, first discussing things that apply to the whole area, and then later discussing specific methods.
I won't lie, this article book is a bit of a slog. You try writing a book about the state of the art of realistic germline engineering in a way that is automatically fun, and then get back to me, ok? But listen: There's lots of pictures, and some of them are good pictures. You could just skim through and look at the pictures, and then read more if there's something you really want to know. If you're looking for mathy stuff, or you just want to hear about the genomic vectoring methods, start here. Also, if you have some sort of oomph you might put into this (such as brainpower or moneypower), or if you're up for a recorded and published conversation, you can message me or email me, and I'll talk you through this stuff. Gmail address: berkeleygenomicsproject
Takeaways
-
The bottleneck to strong human germline genomic engineering is not polygenic scores. The bottleneck also isn't really about making a cell with the genome you want. Rather, the bottleneck is epigenomic correction—making an engineered cell have the right epigenomic state so that it can make a healthy baby.
-
You would be surprised at how soon strong germline engineering can be made technically feasible.
- The scientific and technological precursors to strong human germline genomic engineering are ripening. There are several paths that look fairly feasible.
- Peering through the looking glass at biotechnology, small feats may appear much easier than they really are, but big feats may appear much harder than they really are. It would probably take much less than a Manhattan project to make germline engineering real; the question is more about deciding to do it, rather than whether it can be done.
-
This is an "enchanted field".
- Many advances could be combined with each other to make the methods safer, more powerful, and less expensive.
- That means that efforts are helpful not just in expected value, but with substantial probability. To some extent this is true of science in general. But in germline genomic engineering, different last-mile startups in the area will likely literally synergize, e.g. through licensing, collaboration, or serving as multiple inputs to other groups (see below).
- Many methods have a nice innovation ramp. You could perform a few edits at first, then more and more; or transplant just one chromosome, then several; or do iterated meiotic selection for one or two rounds, then several.
- The innovation ramp, plus the potential for combinations, sets the field up for a sustained ramp-up of energy once it gets started in earnest.
-
The component technologies—reproductive, genomic—need more funding, talent, and project organization. There are lots of things to do, and the existing academic funding and commercial investment landscape is far from efficient with respect to accelerating germline engineering technology. Experts and their projects need more funding to go faster and create more public goods for frontier assisted reproductive technologies. Some other projects that could be created de novo:
- Create a primate research center focused on studying frontier reproductive technologies, e.g. verifying that they produce healthy offspring. Make an atlas of single-cell RNA sequencing data in primate embryonic development.
- More epigenomic sequencing for human reproduction. E.g. characterize the natural range of variation in epigenomic state between single cells within one early-stage human embryo.
- More talent and funding for ovarian follicle culture, which is a likely way forward for making oocytes in vitro.
- Some genomic vectoring methods could likely be applied today or very soon in animals. Testing these methods would give valuable feedback about the techniques and about what happens when you genomically vector strongly according to a PGS in one shot.
- Gather more human phenotype/genotype data, especially for personality traits.
- Develop methods for intact chromosome transplantation. (Three remarkable and little appreciated facts: First, recombinant chromosome selection is a quite strong genomic vectoring method. Second, average chromosome selection with many donors is also quite strong. Third, chromosome selection might largely bypass the epigenomic correction problem, which is a major bottleneck for strong genomic vectoring. It might be infeasible, but for reasons decorrelated from other approaches.)

- Scientists, technologists, investors, grantmakers, parents, policymakers, other stakeholders:
- If you want to help the field along by coordinating, starting projects, or funding work, we'd love to hear from you in DMs or at this gmail address: berkeleygenomicsproject
- Consider joining us at the Reproductive Frontiers Summit 2025.
Disclaimers
I'm not a biologist. Parts of this article attempt to summarize some aspects of a large, complex, changing subarea of biology—the cell biology of human reproduction—as it relates to the possible future technology of human germline engineering. These summaries will necessarily be very incomplete, and unfortunately likely contain errors and confusions. Further, my knowledge is only a few steps from the shallow end, so for example I might say things based on outdated consensus. My hope is that the summaries, while lossy, will help others think about the subarea by pulling lots of threads together into one place and analyzing the basics of how those threads interact with germline genomic engineering.
This article is not intended as an explainer, but rather an attempt to summarize the state of the art for people interested in understanding or contributing to future developments. Unfortunately several of the sections are not independent from the rest of the article, so you might have to jump around and also internet search things.
In trying to find ways to implement strong human germline engineering, my understanding of what's possible, feasible, or easy seems to continually change, even aside from the fact that the field is progressing. There is a "layman's optimism" I've encountered in myself and in others. For example, to my layman's eyes, chromosome selection seems so simple—you just, you know, move the little guys around a bit until they're all together. But any specific plan has big holes in it (micromanipulator? nah, chromosomes are tiny; FACS? that'll probably break the DNA; do it on sperm DNA? it's highly compacted and inaccessible; etc.). Yes, a woman could extract some of her ovary tissue and then grow a myriad of eggs... if she knows someone who knows how to do ovarian culture well, and if ovarian follicle dominance doesn't get in the way too much. Yes, you could edit spermatogonial stem cells and transplant them into testes... but they'd be outcompeted by unedited spermatogonia and die out. I've tried fairly carefully to not overstate the feasibility of methods.
The math about the genomic vectoring power of different methods should be fairly solid, given the assumptions I make. The assumptions that connect the math with actual cells and DNA moving around are shakier; I make many simplifications, some knowingly and some not. The conclusions I give are based on simulations of a simplified abstract model of genomic selection protocols, and is not based on using real DNA sequences of real genomes and operating on those. But I think that the qualitative conclusions should hold fairly well in most cases—e.g. comparisons of strength between different methods, and general ballpark estimates of strength. My hope is to communicate not "this is exactly how powerful these methods are" but rather "a natural first-order estimate says this method is really strong". I think history shows that "surprisingly large first-order estimate plus lots of complications" is, while very far from a sure bet, nevertheless often a very good thing to bet on.
In this article I speak solely on my own behalf.
The entire process of reproductive genomic vectoring (i.e. human germline genomic engineering) is likely to be complicated, especially at first, and hard to foresee. This article is not trying to address everything that would be required for reproductive GV. For example, any reproductive GV protocol must involve several health verifications, e.g. genome sequencing, epigenome sequencing, and morphological normality. This article just addresses the two core elements: genomic vectoring and epigenomic correctness.
Terms
Non-standard terms
Most terms in this article are standard. Some non-standard terms:
- Genomic vectoring (GV)
- Unfortunately this conflicts with "germinal vesicle oocyte". In this article, GV always means genomic vectoring.
- Reproductive genomic vectoring
- Epigenomic correctness (EC)
- Chromosome selection
- Iterated meiotic selection (IMS)
Standard terms
Some standard terms with abbreviations:
- PGS (polygenic score)
- SNP (single nucleotide polymorphism)
- IVG (in vitro gametogenesis)
- IVO (in vitro oogenesis)
- IVS (in vitro spermatogenesis)
- ESC (embryonic stem cell)
- iPSC (induced pluripotent stem cell)
- PGC (primordial germ cell)
- SSC (spermatogonial stem cell)
- Combinatorial abbreviations:
- Before an acronym: i means induced, h means human, m means mouse
- After an acronym: LC means "-like cell"
- So an miPSC is a mouse induced pluripotent stem cell, and an hiPGCLC is a human induced (primordial germ cell)-like cell .
- This is quasi-standard, but to clarify: I use -etic (genetic, epigenetic) to refer to a small number of DNA loci, and -omic (genomic, epigenomic) to refer to genome-wide effects.
Reproductive genomic vectoring
Reproductive genomic vectoring means making a baby who has a genome that was intentionally influenced, rather than solely by the natural reproductive process.
There are many downside risks, both technical and social, to reproductive genomic vectoring. See "Potential perils of germline genomic engineering". I'll address these elsewhere, along with the case in favor of germline engineering.
Genomic vectoring (GV)
Genomic vectoring (GV) means making a cell that contains a genome that has been modified to score highly according to some criterion. The genome could be diploid—a full complement of 46 chromosomes, two of each index, like all non-germline cells in your body; or it could be haploid—23 chromosomes, one of each index, like sperm or eggs.
Once you've genomically vectored a cell, you aren't done. Your GVed cell might just be a generic stem cell. It's not automatic that you can make a healthy baby from any old stem cell.
The epigenomic correctness (EC) problem is the problem of making cells that are epigenomically developmentally competent: they have the right epigenomic states so that they can contribute to growing a healthy baby. Any GV method requires some way of handling the epigenomic correctness problem. See the later sections "Reproductive GV and epigenomic correctness (EC)" and "Methods to handle epigenomic correctness".
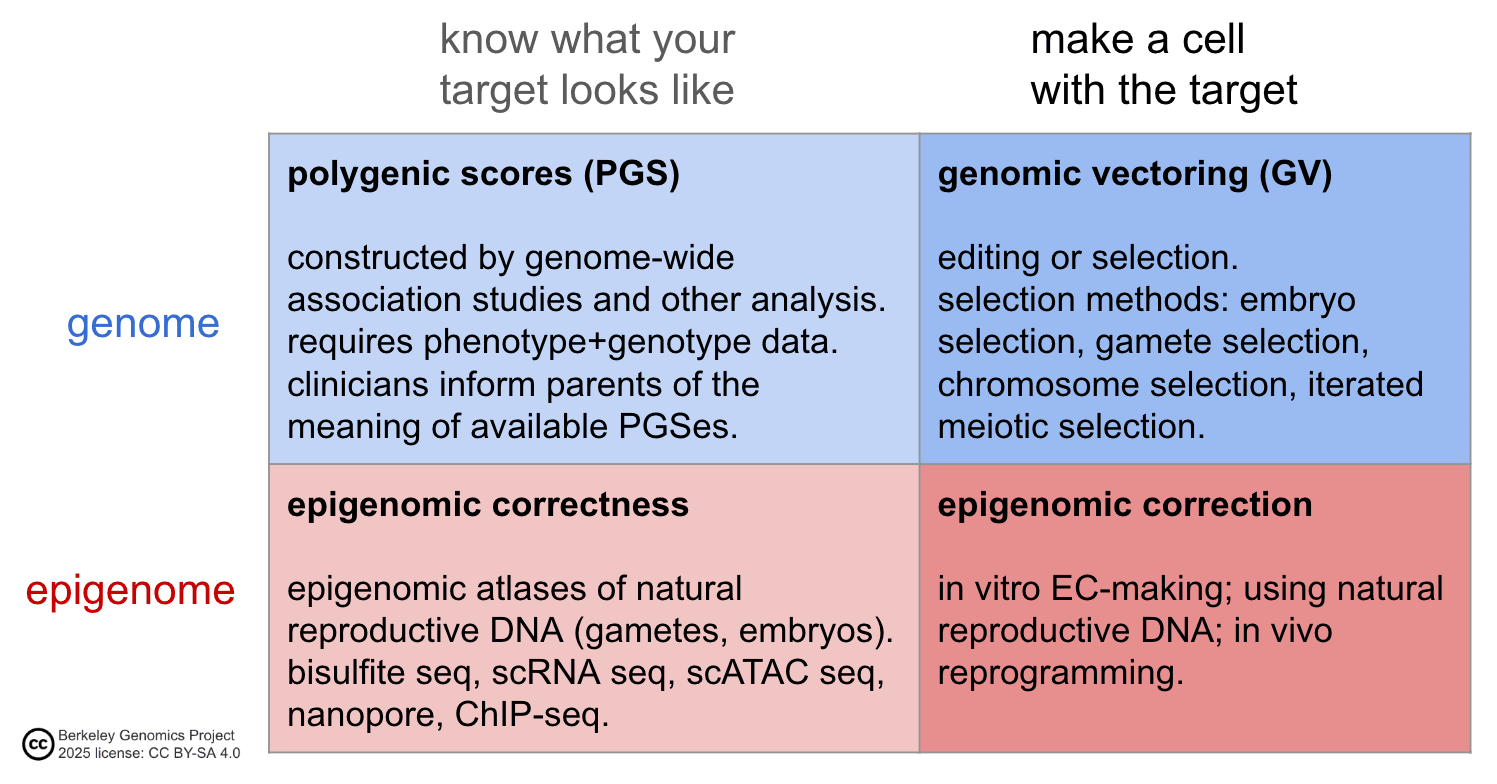
As a view from 30,000 feet, the elements of a method for human germline engineering are to know and make the target genome and epigenome in a cell:
Strong GV and why it matters
By "strong GV", I mean something a little nebulous. The term is meant to include methods that can greatly decrease the risk of several diseases in a future child, or substantially enhance some capacity, e.g. afford 30 IQ points or more. The term is meant to exclude simple embryo selection on normal numbers of embryos, and is meant to exclude editing a few loci.
Strength matters for reproductive genomic vectoring:
-
A stronger GV method can be used to improve the baby's health on more dimensions (decreasing the risk of more diseases). Besides the obvious, decreasing disease risk is good because it helps to protect the baby against potential health problems introduced by the GV method itself.
-
Strong GV removes many difficult tradeoffs between traits. With weak GV, parents have to evaluate whether they prefer to decrease their child's risk of diabetes by an additional 0.5% or to increase their child's expected IQ by a couple points (or something). With strong GV, the question becomes about what genomic foundation do the parents view as most desirable to give their children. Strong GV methods also remove tradeoffs between GV strength and the similarity of the resulting children.
-
GV strength is generally interchangeable with cutting costs. Cutting costs is crucial for making GV technology widely available, in order to have the greatest benefit and to prevent problems with inequality.
-
Strong GV would enable parents to have children with more opportunity for true genius of some flavor—scientific, scholarly, artistic, philosophical, political, communicative, technological, organizational. This would give the next generation high capability for both personal thriving as well as intellectual and altruistic contributions, such as helping humanity navigate the rapidly changing world.
-
If a strong GV can be made safe and soon, then we can more quickly demonstrate the large benefits of the technology. This will more quickly bring the scaled-up accessibility of very beneficial implementations of the technology.
Polygenic scores
Generally, the criterion for vectoring is a polygenic score—a function that predicts a trait from a genome. Genomic vectoring (GV) seeks to make a cell that scores highly according to the PGS, i.e. is predicted to make a baby who has a high degree of the trait/s.
The analyses in this article assume linear PGSes, i.e. PGSes that don't model gene-gene interactions. In practice, the criterion would be a PGS formed as a weighted sum of PGSes for various traits (or maybe a more complicated function of PGSes, such as an intersection of acceptable ranges). This article doesn't discuss what PGSes to use, how to use them, how to get them, how good they are, and what exactly they do and do not mean. In abstracting from those details, this article also doesn't account for the failure of PGSes to fully transfer between ancestry groups; it's worth keeping in mind that more data will be needed to afford the full opportunities of germline engineering to everyone.
IQ is used as a practical touchpoint to understand the power of different GV methods. It's convenient because it's a highly polygenic trait, and there already exist PGSes that correlate with IQ by at least 0.4; and we have some coarse sense of what the trait means. Also, it's a trait that to me seems quite important, in that I want to have clever kids, and I want other parents who want to have clever kids to be able to do so. That said, there are lots of other quite important traits that would be great to have PGSes for, from disease risks and longevity, to personality traits and more specific cognitive capacities (including ones that are also very relevant to intellectual achievement, e.g. courage and curiosity). Also, I have not evaluated the literature on IQ PGSes. If restricting to a PGS that only uses correlations that are actually causal would give a correlation less than .4, then all the estimates in this article about IQ specifically would have to be adjusted down proportionally (just multiply the raw SDs by the true correlation rather than by .4; or multiply the IQ points by ).
See "Embryo Selection For Intelligence", Branwen 2016[1], for more about the genetics of intelligence (and early discussion of GV methods).
Editing and selection
There are two broad types of methods of genomic vectoring (GV):
-
Editing.
- Basically, this means going into the DNA and deleting, adding, or replacing some of the DNA sequence.
- Examples: double-strand break editing; base editing; prime editing; Fanzor editing.
- In more words: editing involves (somehow, e.g. with a viral vector) delivering some molecules (such as CRISPR-Cas9 or another CRISPR system) into a cell. The molecules then cut or nick some nucleotides from the DNA, and/or chemically modify the DNA. Then, some ambient DNA repair machinery makes some other nucleotides replace the ones that were removed or mismatched, maybe with the help of another part of the editor. Thus the DNA gets changed at that location.
-
Selection.
- Basically, this means taking DNA segments that already exist in cells, and then assembling those segments into new combinations in a new cell.
- Examples: (simple) embryo selection; iterated embryo selection; iterated meiotic selection; chromosome selection.
- In more words: selection involves shuffling around chromosomes or large segments of chromosomes. The shuffling methods can be either natural, such as gametogenesis or fertilization, or artificial, such as cell fusion or mechanical chromosome manipulation. By shuffling DNA and using DNA sensing, cells with targeted DNA segments are created and identified.
Other types of genomic vectoring (GV)
Cloning is an edge case in that it targets a genome that already exists. This article doesn't discuss cloning because it has its own ethical problems, and it's not what most people want to do. Also, cloning is generally risky: most cells accumulate genetic damage, and so are dangerous to use to make a baby. Thus cloning would require another genomic vectoring method (as well as major epigenomic correction) to work safely.
Whole genome synthesis is excluded because I don't know about it. Reliably synthesizing tens of millions of base pairs, as would be required to make human chromosomes, is not currently feasible. It also may have disqualifying dual-use risks. If whole genome synthesis becomes feasible, then most of the pathways here are irrelevant. However, full epigenomic correction would be necessary. So, reproductive GV methods that somewhat bypass the epigenomic correction problem would still be front-runners until full epigenomic correction is solved.
Comparing editing and selection
Some broad remarks:
Editing produces out-of-distribution DNA
- Compared to selection, editing takes the resulting DNA more out-of-distribution.
- Selection just stitches together large preexisting segments of DNA. Further, most methods do the stitching in places where natural reproduction might also have done the stitching (by recombination in natural gametogenesis). We can therefore be confident, at the local level of haplotypes (shortish segments of DNA on one chromosome), that genomic selection doesn't produce anything weird or dangerous.
- Editing, on the other hand, cuts DNA at artificial spots. The most efficient editors, base editors, target single nucleotides. These editors thus create novel haplotypes. It's therefore harder to be confident that the results are safe. For example, if you edit a SNP, it could be that there are nearby rare variants that are correlated with the SNP.
- There's at least one theoretical reason for worry: nearby base pairs are in strong linkage disequilibrium, and therefore might be coevolved with each other. If so, it's bad to modify a single base pair in haplotype to be the base pair from haplotype : some variants in haplotype might be good or fine with the allele , but bad with the allele . In other words, the edit pairs with and , which it didn't coevolve with. There would have been very little selection pressure to get rid of -associated variants in and that are bad when paired with , since they don't in fact naturally appear with . (I have heard there may be good reason to think this isn't a problem, but haven't followed up on this. For one thing, the number of such oddities would be quite small in the scheme of things, though also selected for being in important DNA regions.)
Editing is already minimally working technology
- Compared to most selection methods, editing is much more worked-out.
- Except for simple embryo selection, any genomic selection will involve methods that aren't currently known, such as in vitro gametogenesis, in vitro meiosis, or intact chromosome transplantation.
- Genetic editing, on the other hand, is a big industry with rapid progress and lots of existing methods that already work well. There will still be many details to work out, especially with iterated multiplex editing, and there are known and unknown variables that will make iterated editing difficult, but the basic principle is proven.
- However, all strong genome editing methods require some form of epigenomic correction and/or maintenance.
Headwind of mutation
- There's always a headwind of mutation in GV (genomic vectoring) methods.
- Most GV methods, of either kind, involve culturing cells for several generations. Many GV methods also involve doing other operations to cells, such as isolating and passaging single cells to another petri dish, or filtering cells according to some reporter.
- These operations introduce risks of genomic degradation. Mutations are introduced at some slow but steady rate; survival in culture might tend to select for certain mutations; and passaging might further select for mutations.
- For example, a cell being non-sticky is both cancer-associated and also makes it easier to isolate for monoclonal passaging. In lab settings, many stem cell lines carry oncogenic mutations.
- This issue can probably be overcome by dovetailing whole genome sequencing and GV, to continually filter out cells with damaged DNA, but it's a pervasive complicating factor.
- Some methods circumvent the headwind of mutation. Simple embryo selection and sperm chromosome selection probably don't interrupt the natural reproductive process enough to add much mutation.
Fixing de novo mutations
- Both editing and selection should be able to fix de novo mutations coming from the parents.
- Organisms accumulate genetic mutations in their cells as they age; in particular, stored oocytes accumulate mutations (though at a reduced rate) and spermatogonial stem cells accumulate mutations.
- Further, meiosis itself can cause mutations.
- Editing and selection should both be able to fix these. Editing can just undo the change, at least for SNVs or other small changes. Selection can select the undamaged allele. (But selection can't fix damage in a Y-linked locus in male genomes, unless the damage only occurs in some of the Y chromosomes available.)
- I'm not sure which is more efficient though. On the one hand, there may be around a hundred de novos in a given diploid genome assembled from the parents, so selection has a lot to work with. Also, selection can target larger kinds of damage without much additional cost. On the other hand, to fix a specific error, selection has to make a decision about a whole large segment (chromosome or significant fraction of a chromosome), so it seems like more selection power is expended in that toy example.
Bypassing natural integrity checks
- Artificial reproduction might let through more bad mutations and aberrant epigenomic states compared to natural reproduction.
- Gametogenesis and fertilization involve many millions of cells undergoing various selective filters, e.g.: ability of gametogonia to proliferate, ability to appropriately respond to regulatory signals, completing recombination and the rest of meiosis without tripping too many DNA damage detectors, oocyte dominance contests during ovulation, physically passing through reproductive organs, and supporting early embryonic development. Germline cells with bad enough genomic or epigenomic problems don't naturally make it into a conceptus.
- Artificial reproduction would skip some of these steps, removing some of the selection pressure towards integrity. The germline-like cells that would have died out in a harsher in vivo context, but make a conceptus artificially, might have more genomic or epigenomic problems.
Selection makes similar children
- By default, selection GV methods produce children with more genetic overlap than normal siblings.
- Most of the discussion of GV methods in this article focuses on making a single child.
- For people who want to have several children with reproductive GV, if they use a GV method that is a selection method, there would be a tradeoff. If they select fairly strongly according to some PGS, they're likely to make children who are significantly more similar to each other than normal siblings. Selection methods move whole large DNA segments around. If some DNA is anywhere nearby a patch of PGS-high-scoring alleles (like, in the same quarter of a chromosome or something), that DNA will tend to make it into many of the GV-selected cells. So GV-selected cells will tend to share that DNA more often than a random coinflip, as with natural random reproduction.
- This does not apply to editing methods, which change a tiny fraction of DNA (less than one base pair in a million), out of mostly random natural DNA. (However, it's conceivable that some noticeable phenotypic similarity would be induced. For example, if multiple children received all the same high-effect IQ-increasing edits, those edits might have had some specific flavor of effect on cognition, e.g. on personality.)
- I think most of the conclusions about selection methods wouldn't be qualitatively changed too much if the similarity / selection power tradeoff were taken into account. But, likely the GV power would decrease by some amount. The situation is complicated so it's hard to tell exactly how much; there is a lot of variation available in the genome to select from, even just in one person's genome. In particular, strong forms of selection GV would be able to alleviate the tradeoff, by simply promoting different high-scoring segments in different children.
- A simple touchpoint:
- Suppose that a couple wants to have two children using a selection GV method. Suppose also that they want the children to have strictly half genomic overlap, as would two normal (non-twin) children. How much selection power can they get?
- Suppose further for simplicity that we're only focusing on chromosome selection, and we only use the parents's chromosomes. In this case, the answer is: they can get at least half as much selection power as if they just made twins with all the highest-scoring chromosomes.
- The strategy is this: Pick half of the chromosome indices 1—23. For that half, you pick the higher-scoring chromosome from each of the two parents. Both kids will receive a copy of those chromosomes. For the other half of indices, for each parent, you'll randomly pick which kid gets one homolog and which gets the other from that parent. You've used perfect anti-correlation in the latter half of the genome. Thus you concentrate the difference-between-kids chromosomes, and separate them out from the high-scoring-chromosomes.
- You can do better than this by choosing the indices to select on separately for each parent, specifically to find where the score differences between the chromosomes are largest.
- If the parents prefer 75% similarity instead of 50%, they can have 75% of the selection power with the same strategy—overlap on 75% of chromosomes, anti-correlate on the other 25%.
- Many of the estimates in this article about the power of chromosome selection ignore the 23rd chromosome, i.e. we assume we don't select over the 23rd chromosome. Partly that's just for modeling convenience, since the sex chromosome behaves differently than the other chromosomes. But also, it seems strange to imagine parents wanting to have the sex of all their children be the same, just to get a tiny bit more selection power. The difference is less than 5%; see the appendix "Ignoring the 23rd chromosome".
Selection and editing benefit differently from stronger PGSes
-
As the strength of PGSes for target traits increases, selection will directly proportionally increase in power; but editing will have a different curve of increase in power.
- The issue for editing is that it is bottlenecked on number of edits. For a given combined PGS, you pick the top edits to make, where is however many edits you expect to be able to make. Improving one of the PGSes marginally may find lots of weak-effect variants without finding any or many large-effect variants, which wouldn't increase the power of a -edit protocol.
- Selection, on the other hand, takes advantage of all the information across the whole genome at the same time, so to speak. If you simultaneously improve the correlation all of your component PGSes with their target traits by a factor of , you then also multiply by the correlation of your combined PGS with the combined target trait. Selection GV methods, without changing the protocol power, would then have their final effects multiplied by as well.
- The same applies for adding more PGSes (that is, mixing in a newly constructed PGS for a previously untargeted trait). Both editing and selection will get more powerful (in terms of total effects on the new combined score), but selection will improve much more.
-
However, the scale situation is not necessarily so bad for editing.
- In some regime, including right now, there's significant uncertainty about which exact SNP within a segment of 5ish SNPs is the actual causal SNP[2]. Improved PGSes should narrow that down, which should substantially improve the expected effects. Selection would benefit little or not at all from more precise causal information (at least within the same population as the source of the PGS): selection already captures causal variants by selecting for larger DNA segments, which contain the causal variant and its nearby correlates.
- For many/most disease traits, the gains for any method are rapidly diminishing. The first few changes in disease alleles, starting with a normal genome, will make much more difference in absolute disease probability, compared to changes made to an already especially low-risk genome. E.g. a move from 3 SD to 5 SD low risk for some disease might represent only a .0001 difference in disease probability, but a move from -1 SD to 1 SD might be a .05 difference, or something. (In contrast, returns for IQ variants don't diminish nearly as quickly.)
- There may be many rare, high-effect variants, as is the case for some traits. Improved PGSes would progressively discover these, thus unlocking marginally higher-effect edits. However, these rare variants would plausibly come with an increased risk of unintended effects, especially if they have a strong enough impact on the target trait that they make it into the top over more common variants.
Selection distributes Euclideanly, editing distributes Hammingly
-
Selection distributes SDs of selection power Euclideanly to component PGSes.
- See https://tsvibt.blogspot.com/2022/08/the-power-of-selection.html#6-selecting-for-multiple-scores.
- This means that if you can select a genome so that it is standard deviations (SDs) extreme, you get points. Then you distribute those points to different traits (for which you have PGSes). If you give points to a trait, that trait will be SDs extreme, where is the correlation coefficient of the PGS with the trait.
- For example, say you select a genome to be 5 SDs extreme. You get 25 points. You could allot them all to IQ, and then you'd get IQ points, because we have a .4 PGS for IQ. Or you could put 16 points into IQ, and use up the rest of your points by putting 3 points into each of 3 health traits. Then you'd get IQ points, and SDs on each of the health traits (if, say, they each have a .3 PGS).
-
Editing distributes editing power (quasi-)linearly.
- Editing moves the genome around in genome-space, which is a sort of Hamming space. At a given level of development, an editing protocol can take some number of steps—i.e. make some number of edits.
- With editing, the question is: what are the most trait-positive variants that you know about, how many aren't already in the genome you're editing, and how many edits can you make given the mechanics of cell culture and editing molecules.
- Given a number of edits and multiple target PGSes, you have to simply apportion each edit-slot to one of PGSes. This is "linear" distribution.
- (However, for each trait, some edits will have a larger SD effect on that trait than others. So the literal total number of SDs (which is not an important metric) is not fixed even with a fixed edit count; hence "quasi-linear".)
Selection beats editing, then editing beats selection
- Selection is stronger than editing when both are weak; editing is stronger when both are very strong.
- In other words: A weak version of selection will have a greater effect on traits compared to a weak version of editing. On the other hand, strong editing has a greater effect than strong selection.
- Examples of weak/strong versions of selection:
- chromosome selection on one/many donors,
- iterated recombinant selection with few/many iterations,
- simple embryo selection with an ordinary/gigantic number of eggs.
- Weak editing is modifying up to a couple hundred loci; strong editing is editing many hundreds or thousands of loci. (In the limit, editing is as powerful as whole genome synthesis.)
- For polygenic traits such as IQ and many health, longevity, and other cognitive traits, each genetic variant has a very small effect on the trait. For example, all or very nearly all IQ variants will have less than a .5 IQ point effect.
- Weak selection is stronger than weak editing. Weak selection can weakly harness the power of the entire suite of available PGSes. Weak editing can only edit, say, the top few dozen most effective variants, and has to ignore all the others.
- Strong editing is stronger than strong selection for two reasons. First, strong editing can add variants that aren't available in the parents's genomes, but that are known to be good because other people have them. Second, editing can, in principle, add variants that no human has, but that are believed to be good for some other reason (e.g., a variant coding region improves a protein's efficiency; or the variant is tested in chimps and improves something).
- In any case, strong versions of either type of vectoring are sufficient to get enormous benefits.
Editing can reach more places than selection
- In general, editing can be applied to cells in more contexts than selection methods.
- Editing can be done by delivering some molecules to some cells, wherever those cells are. For example, it's even possible to edit adult tissue in vivo, though with difficulty and with low efficiency. A practical example is that one might be able to edit gametogonia that inhabit gonadal tissue in vitro.
- Most selection methods, on the other hand, require filtering cells and DNA. E.g. chromosome selection requires moving specific chromosomes between specific cells; iterated recombinant selection requires fusing specific haploid cells.
The ceiling of safe vectoring
- Some GV methods that are likely to be feasible would hit the upper limit of what would be safe.
- Generally speaking, no trait should be pushed too far. Some examples:
- IQ, if pushed too high, would produce severe physiological problems. See the bullet point "Traits outside the regime of adaptedness" here.
- Health traits would likewise have unknown physiological effects if pushed too far.
- Personality traits are likely bad at extremes. As a possible example: slightly low conscientiousness might produce creativity, and slightly high conscientiousness might produce diligence and carefulness; but very low conscientiousness might produce dysfunctional erraticness and psychopathy, and very high conscientiousness might produce neuroticism / OCD. Slightly low agreeableness might produce leaders and independent thinkers, and slightly high agreeableness might produce good team players and caregivers; but very low agreeableness might produce violent psychopathy[3], and very high agreeableness might produce exploitability or subservience.
- Editing, if scaled up enough, would hit limits. Likewise chromosome selection with many parents and iterated meiotic selection would also hit limits.
- Generally speaking, no trait should be pushed too far. Some examples:
Many GV methods synergize
Many methods for genomic vectoring (making a cell with a genome that's been nudged in some direction) can be combined with each other to increase power and/or decrease costs. There are many possible combinations of different methods, and until some GV technologies are ready to be applied it doesn't matter exactly how well various combinations work. To give some main examples:
- Chromosome transplantation would enhance most GV methods.
- For most methods that operate on multiple cell lines, chromosome transplantation would allow gains in chromosomes from different cells to be aggregated.
- E.g. if you iteratedly CRISPR edit some cells, you may have chromosomes in multiple different cells that successfully got the most target edits, and the least DNA damage. It would be nice to then just move those chromosomes together into one cell.
- E.g. see the appendix "The costs of iterated meiotic selection" on how chromosome selection makes IMS less costly / more powerful by gathering together chromosomes that have target crossovers.
- E.g. if editing isn't available, chromosome transplantation would enable preventing monogenic homozygous diseases by replacing one chromosome from a donor without the disease allele.
- Editing would enhance most GV methods.
- For most methods that operate on culturable cell lines (so that you can sequence some cells to check for damage), editing would allow spot-correcting any damage, whether due to parental de novos, culturing, or abnormal cellular processes (e.g. inducing meiosis might cause DNA damage).
- For selection methods, editing can introduce variants not present in the starting population of cells. So it can prevent homozygous disease alleles from being passed on, and it can enhance traits beyond what would be achievable just from the starting population, e.g. by editing in known helpful rare-ish variants.
- Embryo selection can be combined with any method that creates a viable haploid gamete-like cell, effectively selecting the complementary gamete as well.
- DNA sensing can help any selection method.
GV synergy has two very auspicious consequences:
- If a GV method can be made to work, it's unlikely to be a dead end. Even if a method is delayed, can't produce much GV on its own, or is costly, there's a good chance it will still be useful as an admixture to another successful GV method. This somewhat decreases the risk a research project faces that its products will not be interesting or useful.
- Once any nontrivial GV method is working, it's likely that there could be a large, fast increase in the strength of the strongest combined GV method. As mentioned above, strength is important for protecting the baby from possible side-effects of the GV method, for making the technology widely available, and for granting greatly enhanced capacities.
GV synergy also highlights the key bottleneck in reproductive GV: dealing with the epigenomic correctness problem. There are several synergistic paths to GV which could work on their own or together, so I'm not very concerned that we won't have a good combined GV method. However, any full reproductive GV method requires solving the EC problem, whether with an EC-making (epigenomic correction) method or by bypassing the EC problem (e.g. using natural reproductive DNA).
Reproductive GV and epigenomic correctness (EC)
Reproductive genomic vectoring means making a cell with a vectored genome, and then making a healthy human baby from that cell.
The epigenomic correctness problem
To reliably produce a healthy human baby from a cell, you must make sure that the cell has the right epigenomic state. The chemical markers on and around the DNA—e.g. CpG methylations and histone modifications—should look like the markers on natural reproductive DNA (eggs, sperm, zygotes).
Suppose you just take the nucleus from a somatic cell, or even a stem cell, and stick the nucleus into an oocyte and try to implant it. There's a high chance it won't grow, or if it does grow, there's a high chance it will miscarry or be stillborn, or if it isn't miscarried or stillborn, there's a high chance the resulting baby will be sick and deformed. That's because a random cell won't have the correct epigenomic state. This is the "epigenomic correctness" problem. (Or "epigenomic competence".)
In natural reproduction, gametogenesis produces gametes (sperm or eggs). The early stages of germline development leading up to gametogenesis involve wiping clean the epigenomic state of the stem cells that will become gametes. Genome-wide reprogramming happens in both sperm and eggs, but there are also sex-specific epigenetic imprints: spermatogenesis produces spermatozoa (sperm) with paternal epigenetic imprinting, and oogenesis produces oocytes (eggs) with maternal epigenetic imprinting. These sex-linked epigenomic imprints are different between the sperm and the egg, and are necessary for healthy growth of the fetus[4]. An illustration:
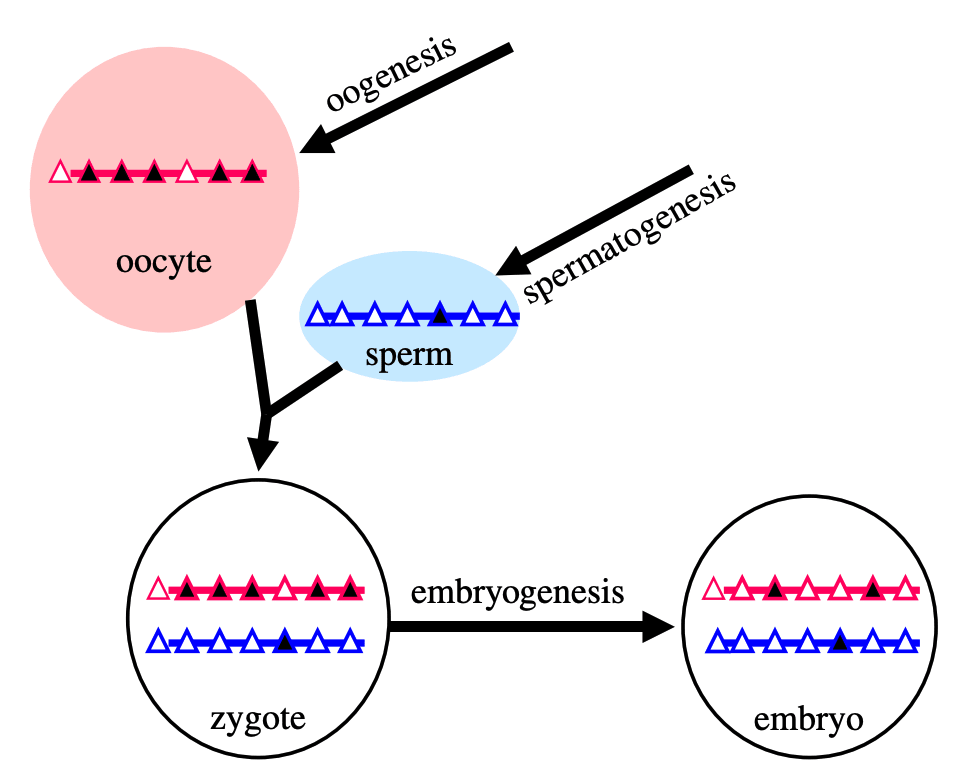
[(Figure 1 from Kelsey and Feil (2013)[5].)]
The triangles represent CpG islands in the genome; the black ones are methylated.
It's not known exactly what imprints are necessary or sufficient for healthy development. Some research has epigenomically sequenced human gametes and embryos[6], but we don't have a full picture (e.g., the cited paper doesn't look at histone modifications). It's probably something like several dozen to a couple hundred epigenetic marks across the sperm and oocyte genomes. (Over a hundred sex-linked differences are known, but many are controlled by others and therefore are not independently necessary.) Knowing this information is crucial for most reproductive GV methods. We'd also want detailed information on the epigenetics of fetal development so that we can tell if something is wrong. There hasn't yet been a definitive "gold standard" agreed on for what should count as demonstrating epigenetic safety for making human babies. Some of the main elements would be multi-omic similarity to natural gametes, safety demonstrations in animal models, and morphologically and multi-omically normal embryonic development up to 14 days.
There should be more funding for epigenomic sequencing of germline cells:
- Natural germline cells. Tissue from ovaries and testes from different points in the lifecycle, taken from non-human primates or from humans if ethical (e.g. using tissue from miscarried fetuses with informed consent of the parents). Primordial germ cells, gametogonia, gametocytes, gametes, as well as support tissue (gonadal cells such as granulosa cells and Sertoli cells).
- Especially, normal healthy gametes (sperm and eggs).
- Early embryonic tissue in non-human primates, to get a clearer picture of what healthy development looks like epigenomically.
Broadly speaking, methylations comprise most of the important sex-linked epigenetic imprinting marks, while histone modifications are at least to a large extent downstream of methylations. In sperm, most (around 90%) of the histones are replaced by protamines for tighter packing. However, at least some of the remaining sperm histones at paternally expressed genes retain modifications, so we definitely cannot rule out that some histone modifications are a necessary element of healthy paternal DNA[7]. Tanaka and Watanabe (2023) suggest that the main problem with ROSI is that round spermatids have too many histones that haven't yet been replaced with protamines[8]. In trying to clone macaques, Liu et al. (2018) found that histone modifications were partly determinative of cloning success, so it is possible for histone modifications to matter a lot (though this effect could go away with otherwise normal methylations)[9].
It's not known how much loss of imprinting occurs naturally in somatic cells, or how much would occur as the result of operations involved in GV methods (such as inducing pluripotency with Yamanaka factors, inducing naivety with super-SOX, culturing in vitro, editing, or mechanical manipulation).
It's not known how to take a non-reproductive cell, and then correct its epigenomic state so that it can be a viable gamete or zygote (1-cell embryo). Such an epigenomic correction (EC-making) method would be one kind of solution to the EC problem.
Applying reproductive GV in humans would require a way to ensure epigenomic correctness with pretty high confidence, before making the first baby. However, there are several strong GV methods, such as iterated CRISPR editing or chromosome selection via whole cell fusion, that could be tried soon in plants, mice, or other animals.
Sex-linked epigenetic imprinting isn't the only aspect of the on-DNA epigenomic state that's needed for healthy development. In the early embryo, for example, the DNA has to be broadly demethylated like a natural naive ESC, so that embryonic cells can differentiate into all the tissues of the conceptus. Since broad DNA demethylation occurs naturally, given the environment provided by the egg, we may or may not have to worry about it specifically.
There are also other characteristics of sperm and eggs, or of zygotes, that are important for development, besides on-DNA epigenetic marks. These will be discussed briefly in the later sections on in vitro gametogenesis. The task of epigenomic correction is more narrow: we just need to get the DNA itself in a developmentally competent state, and then if necessary we can use a donor egg and/or sperm to give the rest of the needed support for fertilization and development.
The near-miss hazard of epigenomic correction
Do we actually need to have the right epigenomic state? Organic life is robust, it self-corrects. What if we just put the DNA we want into an oocyte (after getting rid of the DNA already there) and then tell it to grow?
That method is called SCNT—somatic cell nuclear transfer, a.k.a. cloning—and it's how the first cloned mammal was created. In 1997, Wilmut et al. announced they had made a lamb, number 6LL3, from an adult sheep mammary gland cell using SCNT[10]. (That very 6LL3 grew up to be none other than Dolly the sheep.)

[(Figure 2 from Wilmut et al. 1997.)]
Dolly the sheep had 276 siblings who didn't make it. Of the 277 mammary epithelium cells that Wilmut et al. did SCNT to, 29 grew to the blastocyst stage and were transferred into recipient ewes; only one was born. (They also transferred embryos derived by SCNT from embryonic stem cells and from fetal cells; those had a better survival rate, though still bad.) That should be alarming; something is at least quite unnatural about this process.
But maybe it's fine. More than 10% of normal pregnancies miscarry anyway; maybe, as along as a fetus grows for the full term, it'll be fine? Could we simply do SCNT with adult human cells? Perhaps after editing them? No. Don't do that.
Just because a fetus is born alive, does not mean it's healthy. There are plenty of possible developmental abnormalities that don't kill a fetus, but that do severely affect the health or lifespan of the offspring. Observations of 1000 cloned dogs show substantial fractions of them have significant developmental abnormalities such as cleft palate and muscular hypermyotrophy[11]. In cattle, the situation is not good: "On average, 42% of cloned calves died between delivery and 150 days of life; the most common abnormalities were: enlarged umbilical cord (37%), respiratory problems (19%), calves depressed/prolonged recumbency (20%) and contracted flexor tendons (21%)."[12]. Survival and deformity rates vary by species, by cell source, and by IVF method. But it's a minefield, and humans definitely do not have the least finicky mammalian reproductive epigenetics.
Part of the reason SCNT doesn't work to reliably make a healthy offspring is that sex-specific epigenetic imprinting is required for healthy development, even if it's not strictly required for minimal viable development[4:1]. The donor oocyte has enough material (e.g. transcription factors) in the cytoplasm that it can sometimes nudge the transferred DNA enough to sort of muddle through development. But many epigenetic marks will still be missing in cells from adults, having degraded during growth, and development is very prone to be abnormal. Optimizing your protocol to increase efficiency—to make more artificial zygotes make it to a live birth—does not necessarily decrease your rate of deformities nearly enough.
That's the near-miss hazard of epigenomic correction. You correct the epigenomic state enough to make a live birth, but not enough for reliable healthy development. Not good.
I'm going to keep giving examples so you know I'm not making this up.
The most direct observation we can make about imprinting problems is uniparental disomy, explained to me by Tassilo Neubauer. In uniparental disomy, a child inherits two chromosomes of the same index from one parent. (In contrast to trisomy, the chromosome of that index from the other parent is not present for some reason.) Depending on the chromosome index, many people with disomy are fine, but many others have serious health issues. Some of this is due to homozyosity, if the chromosomes are identical; but some is due to the incorrect imprinting of the uniparental chromosomes (maternal-maternal or paternal-paternal). Read more at the UPD database[13] and in Neubauer's short review [LW · GW][14]. For a cell with not just one, but all chromosomes incorrectly imprinted, the effects would be lethal, but if not lethal then quite bad.
Nayernia et al. (2006) derived mouse haploid sperm-like cells from embryonic stem cells in vitro. Then they used those sperm-like cells to fertilize mouse oocytes and implant the resulting embryos. The ones that were born alive were over- or under-grown, and died 5 days to 5 months after birth. They confirmed that the experimental mice had abnormal methylation patterns in sex-linked imprinting regions, specific to whether they were over- or under-grown[15].
Recently, Li et al. (2025) made bi-paternal mice by making genetic edits to tweak the expression of regions that would normally be controlled by the correct imprinting. This worked, in that many abnormalities were prevented... but the mice that were born were sick, and almost all died[16]. Earlier, Li et al. (2016) had done a similar thing to make bi-maternal mice. The research process involved creating, along the way, severely undergrown bi-maternal mice[17].
Mitalipov et al. (2002) applied SCNT to rhesus macaques, making embryos from blastomeres—very early embryonic cells, which ought to be fairly epigenetically normal. Of 30 embryos transferred into 11 monkeys, only 1 pregnancy resulted. It grew to term, but was stillborn[18]. The authors guess the fetus died of asphyxiation by the umbilical cord, but one wonders if that was induced by some developmental problem caused by epigenetic abnormality; asphyxiation stillbirths are rare, and recall that cloned cattle frequently have enlarged umbilical cords[12:1]. Since the zygote genome is quickly reprogrammed (almost entirely demethylated) soon after fertilization, it stands to reason that there could be a difference in SCNTed ESCs that has significant consequences. For example, chromatin condensation of the oocyte genome, which would be messed up by ESC SCNT, might be important for early development[19].
Liu et al. (2018) cloned cynomolgus macaques with SCNT[9:1]. They used a treatment that altered the histone modifications in the DNA to be cloned. This greatly improved the success rate, and they made two apparently healthy offspring from fetal fibroblast cells. But they also tried the same method using somatic cells from an adult macaque. Of the few pregnancies that took, 2 miscarried late-term, and 2 were born alive. Quoting:
Infant A showed normal head circumference but impaired body development at birth and died 3 hr later due to apparent respiratory failure. Infant B had apparent normal head and body development and showed normal breathing and food and water intake but died 30 hr later with respiratory failure (see Data S4).
In humans, epigenetic abnormalities have been somehow associated with serious disorders and cancer[20].
So that's why you can't just make a human baby without knowing what you're doing: You stand a high risk of making a baby with developmental abnormalities that weren't severe enough to abort the fetus, but are severe enough that the child is suffering. If for some reason the moral consequences of that aren't enough to dissuade you, consider that other people would ban you and your children and your children's children and your artificial children and any similar research for 1000 years.
Going from 0.01 to 0.99
Reproductive genomic vectoring involves composing multiple biotic processes together: gene editing and repair, mitosis and meiosis, folliculogenesis, gametogenesis, induction of stem cell states, embryogenesis.
Biotic processes—cell division, gametogenesis, embryogenesis, up- and down-regulation of genes, cell differentiation—are both robust and noisy.
They're robust, in that even if not every cellular process goes exactly according to the evolutionary design, the end result may be almost as good, or even exactly as good. Error correcting mechanisms such as DNA repair, methylation maintenance, self-perpetuating gene regulatory network states, and homeostatic feedback in general, can bring and keep a cell on track (probably, approximately), as long as the cell hasn't been too extremely perturbed.
Biotic processes are also noisy at every step. DNA copying is imperfect, chromosome synapsis is imperfect, DNA breakage repair is imperfect, and so on.
Because of the noise and the self-correction, surprising things happen. Embryonic stem cells cultured in a certain way seem to, in some small fraction, spontaneously undergo meiosis, despite the absence of most of the preconditions for normal gametogenesis. Somatic cells transplanted into enucleated oocytes will, in some small fraction, develop into healthy offspring. This means that with many challenges, we don't exactly start at 0, but rather we start at 0.01: We can do them, but only rarely, or perhaps slowly and at high cost, and with results whose quality is both poor and unknown.
In general, starting at 0.01 is not good enough:
-
A main reason, as discussed in the previous section, is that for some processes, near-misses are very costly.
-
Importantly, the development and safety validation of the technology involves lots of experimental iterations. If the inner loop of the experiments—the biotic processes that you have to recapitulate frequently and in great numbers—is slow and costly, you get much less rapid feedback. It's also harder to compose multiple methods together if they're unreliable, so it's harder to get especially informative end-to-end feedback. And it is harder to share and replicate methods.
-
Also, ultimately we want the whole integrated protocol to be inexpensive and scalable as well as knowably, consistently safe. Inexpensive scalable methods would support bringing reproductive genomic vectoring to more people, and make a stronger case that reproductive GV won't increase inequality due to differential access.
-
Errors may add up.
- For example, a process for creating ovarian follicles might produce medium-quality primordial follicles, that sort of resemble natural primordial follicles. But then if you use the artificial follicles to mature some oocytes, you produce very low-quality oocyte-like cells, because the follicles can't really give the oocytes all the support they need.
-
Poor efficiency
addsmultiplies up.- If you can make 1% of your iPSCs into fully competent mature oocytes, you can eat the cost of that low efficiency. But if only .5% of your immature oocytes become mature oocytes, and only .5% of your artificial mature oocytes can be fertilized and start growing, and only 1% of those embryos can be implanted, then your costs for the full process would be... crunches numbers... "really big".
- For some processes, poor efficiency can be compensated for by amplifying the desired cells at each stage, e.g. by filtering and proliferating cells. But this adds time and complexity costs, and doesn't work for everything (e.g. you can't straightforwardly proliferate oocytes).
So we can't be satisfied with 0.01. We have to get much closer to 1.
However, none of the above reasons demand getting to literally 1. Because natural biotic processes are noisy, our bar for quality does not have to be 100%. Babies born naturally have birth defects at a rate greater than 1%, and natural pregnancies miscarry at a rate greater than 10%. Assisted reproductive technologies should have a higher bar for safety, but by no means should they be required to produce 100% perfect results. Once they are known to improve over the other options, they should be available. We don't need to get to 1, but more like 0.99 (not speaking precisely).
So in this subarea of biology, we're not trying to go from 0 to 1, but more like from 0.01 to 0.99.
Biology's *scopes problem
Biology has had a *scopes problem[21]. Cells are tiny and numerous, and their contents are tinier and more numerous, so it's hard to know what all is going on between and inside cells.

[(I don't know if this is how this meme works but I don't care and you can't stop me.)]
This means that cell types, such as pluripotent stem cells or primordial germ cells or gametes or gametogonia, are not fully characterized; we can't say exactly or fully what it means for a cell to be that type of cell. Also, it's just expensive to measure large amounts of information about cells. Instead scientists use a few markers associated with biotic processes to identify cells during experiments, such as the SYCP3 protein for meiosis, and then later take more costly comprehensive measurements to confirm results.
Measurement tech is developing quickly. E.g. DNA sequencing at scale has only been around for about 15 years, and single-cell RNA sequencing has spread in the past 10 years. Epigenomic sequencing has followed suit (e.g. bisulfite methylation sequencing)[22]. But these technologies are still far from ideal lenses; for example, the standard method for bisulfite sequencing requires applying the bisulfite treatment before amplification (because amplification by default produces unmethylated DNA), and it involves heating the DNA and therefore destroying much of it, so it has very poor coverage for single-cell sequencing. We coarsely point at types of cells by their physiological function, but we haven't yet carefully determined the full meaning of the categories.
Furthermore, many cells are difficult or infeasible to access. Many cells are embedded deep inside 3D tissues, maybe mixed in with an overwhelming majority of cells of some other type. Also, many tissues are either expensive, illegal, or unethical to access, most importantly tissue from humans such as fetal tissue or tissue from a living human's gonads. These cells and tissues are obviously much harder to characterize—we don't know what they look like naturally because we can only study a few examples (e.g. tissue from aborted fetuses or ovary tissue extracted from a woman undergoing cancer treatment for fertility preservation).
All of this means that terms like "secondary oocyte" or "spermatogonium" or "embryonic stem cell" are not 100% perfectly specified terms that point to a comprehensive catalog of known functional behaviors and internal states. Rather, they're phenomenological terms, as in "when transplanted into a conceptus, this kind of cell's descendants can contribute to any fetal tissues but not to the placenta, and maybe we have some noisy mixed RNA-seq data that somewhat characterizes some portion of the gene activity of this kind of cell". So a claim like "we created oocytes / oocyte-like cells" does not necessarily mean "we created cells that are fully competent to contribute to normal healthy embryonic development", and even if we did create fully competent cells we wouldn't be able to fully tell that we'd done so.
Methods to handle epigenomic correctness
There are two basic ways to handle epigenomic correctness:
- Somehow make your cell have the right epigenomic state. These are called "epigenomic correction" / "EC-making" methods.
- Let nature do the work of setting up the right epigenomic state, and do GV in a way that doesn't mess up nature's work. These are called "EC bypass" methods.
The following subsections describe the main types of methods to make cells that satisfy epigenomic correctness. There may be others I'm not aware of. Keep in mind that whether or not a method works for making cells that are actually epigenomically competent to reliably make a healthy human baby, there's a whole separate issue of knowing that it does so, and demonstrating that fact to scientists, regulators, and parents. That issue requires other research such as getting a clearer picture of epigenomically correct states, and validating EC-making methods in animal models to a high enough degree of reliability.
My beliefs about this area are still in flux, so take my claims with salt.
Takeaways:
- Full epigenomic correction in vitro—establishing full maternal imprinting or paternal imprinting—has not been achieved in humans. It also probably hasn't been achieved in mice, in the full sense of getting all the imprinting to look natural.
- Workable in vitro gametogenesis has been achieved in mice: We can make healthy mouse offspring from artificial gametes. But those methods won't translate very well to humans because they use tissue taken from mice, and they don't necessarily meet a high bar of epigenomic correctness.
- The robust way to accelerate the field:
- Fund and do research that builds multi-omic single-cell atlases of human and primate reproduction. Only in the past 5 or so years has it even been possible to do so, because the technology was only recently developed[22:1].
- Fund and create a primate research center that is able to support tests of novel assisted reproductive technology.
- Instead of full EC-making, it may be easier to piggyback on natural gametogenesis. Chromosome selection on gamete DNA might be feasible, would bypass the EC problem, and would be a strong GV method. Iterated CRISPR editing SSCs followed by in vivo transplantation would be a medium-strength GV method.
In vitro gametogenesis (IVG)
Gametes are haploid cells (23 chromosomes, one of each index) that combine to form an embryo; male gametes are sperm (i.e. spermatozoa), female gametes are eggs (i.e. mature oocytes). Gametogenesis is the process whereby stem cells differentiate into gametes. In the natural lifecycle, the germline develops from early on in embryonic growth, culminating in gametogenesis in gonads (adult testicles, or fetal and adult ovaries).
In vitro gametogenesis (IVG) would reconstitute this process in the lab, making artificial sperm (in vitro spermatogenesis, IVS) or artificial eggs (in vitro oogenesis, IVO). IVG would allow us to artificially make sperm or eggs from stem cells that aren't just the natural germline stem cells that are native to humans.
Takeaways:
- Minimum viable IVO and IVS have both been achieved in mice, albeit using methods that wouldn't scale in humans because they use gonadal tissue extracted from organisms.
- Meiosis has been achieved, to some extent, in human male germline-cell-like cells. It hasn't been achieved in female germline-cell-like cells.
- Most research in human IVG so far uses gonadal tissue, so it wouldn't scale.
- Neither paternal EC-making nor maternal EC-making has been achieved in human cells in vitro.
- We need more sequencing data from natural gametogenesis.
- E.g. scRNA-seq, scATAC-seq, scChIP-seq, and bisulfite seq atlases from human gonadal tissue. See e.g. [23] [24] [25] [26].
- We need to know what natural gametes look like, so we know what the results of IVG should look like.
- If we knew what natural gonadal cells—e.g. Sertoli cells, granulosa cells, thecal cells, and germline cells at various stages—normally look like in terms of gene expression, then we'd be able to coax iPSCs to behave likewise. Thus, we'd progress through germline development, and also make gonadal organoids able to support natural gametogenesis.
- This might just work straightforwardly. IVO, and especially IVS, might or might not also need 3D culture methods. Testes in particular have 3D structure that supports sequential steps of development.
- Better methods for culturing gonadal tissue, over long periods and with high quality, would give us a way to cross the EC-making gap that could be accessible sooner than fully artificial end-to-end IVG.
Gametogenesis, and the research about it, is complex, and I am nowhere near to being an expert. The following subsections summarize some main points. For more reliable and complete information, see the reviews by Saitou and Miyauchi (2016)[27], Saitou and Hayashi (2021)[28], Tanaka and Watanabe (2023)[8:1], Robinson et al. (2023)[29], and the other citations from this section. In what follows, I'll paint with a broad brush, glossing over very many potentially important details and distinctions.
The basic elements of gametogenesis
The idea of IVG (in vitro gametogenesis) is to take some stem cells in a petri dish, and make them go through the cellular processes that happen in natural gametogenesis. You use chemicals (culture media, transcription factors, cytokines, gene editors) and surrounding cells to activate and support those cellular processes. Here are the three elements of gametogenesis, which are the changes that a stem cell should undergo to become a gamete:
- make the needed epigenomic changes to the cell's DNA: a. general germ cell epigenomic reprogramming, first broadly wiping state and later silencing most of the genome; b. and sex-specific epigenetic imprinting at several dozen control sites;
- perform meiosis, which makes a haploid cell with 23 chromosomes (one of each index) from a diploid cell with 46 chromosomes (two of each index) through recombination;
- and make the cell develop, through sex-specific morphological and cytoplasmic changes.
The most important element for EC-making is 1., epigenomic correction. Unfortunately it's not very well understood, so I don't have a nice picture.
Meiosis looks like this:
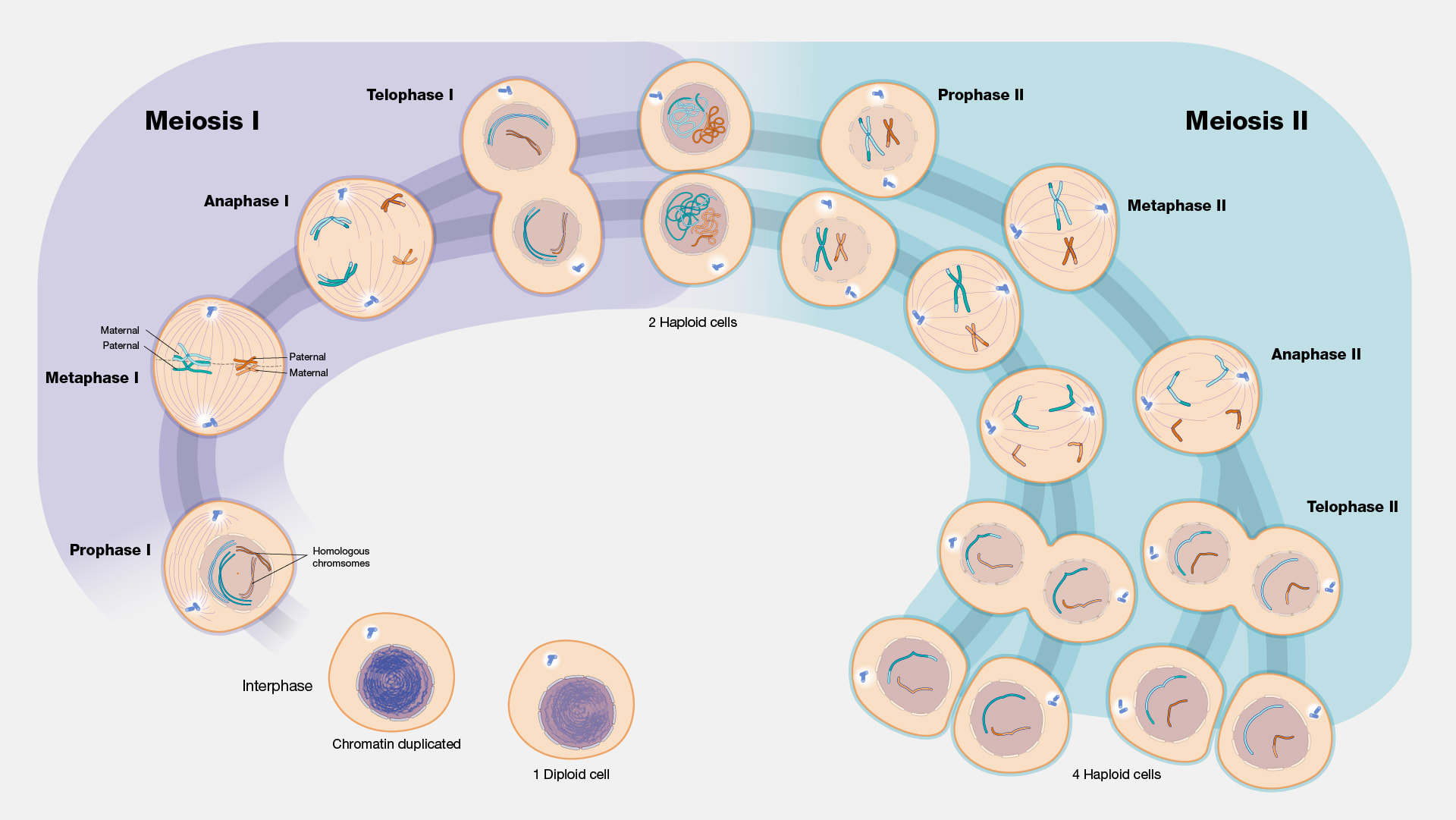
[(Diagram from Gilchrist[30].)]
Here's what mouse and human germline development, including gametogenesis, looks like:
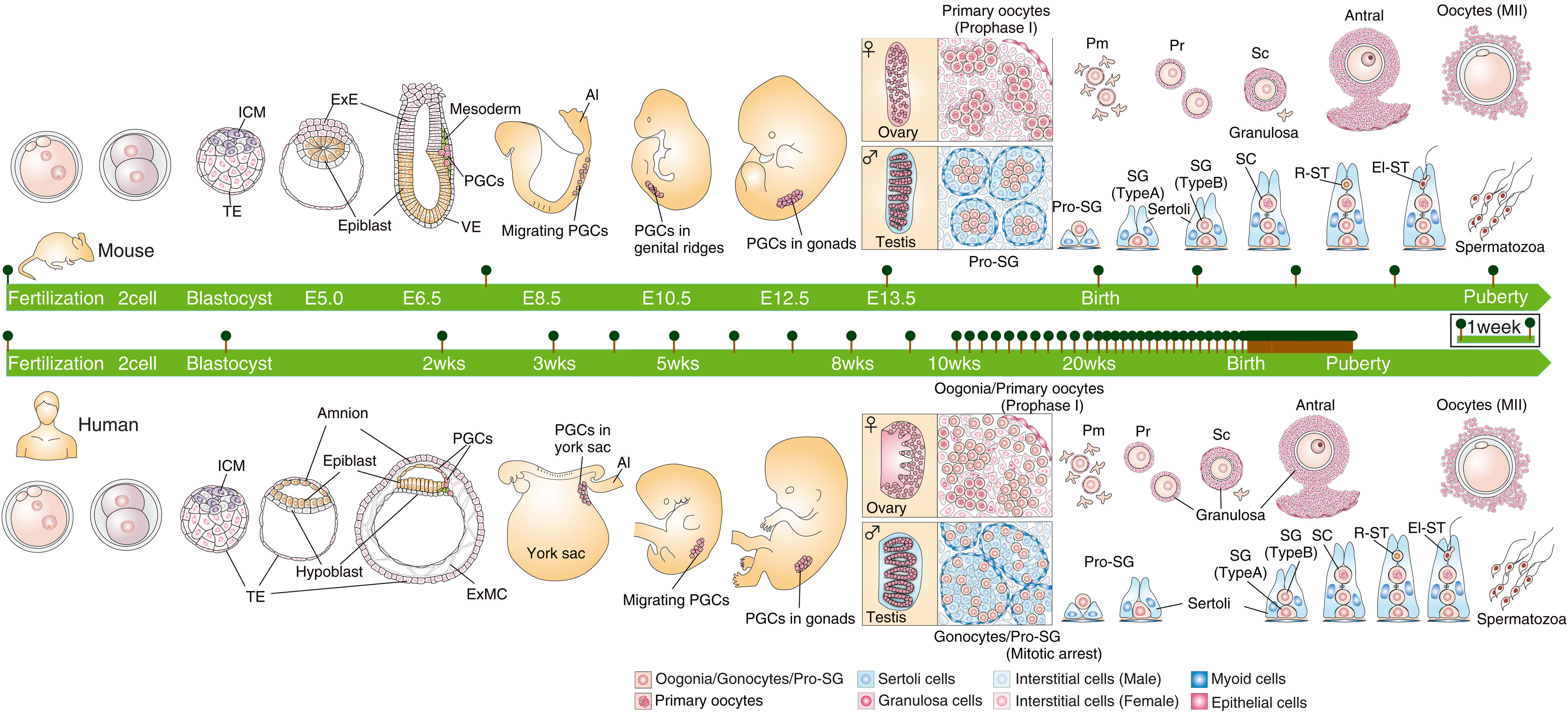
[(Figure 1 from Saitou and Hayashi (2021)[28:1].)]
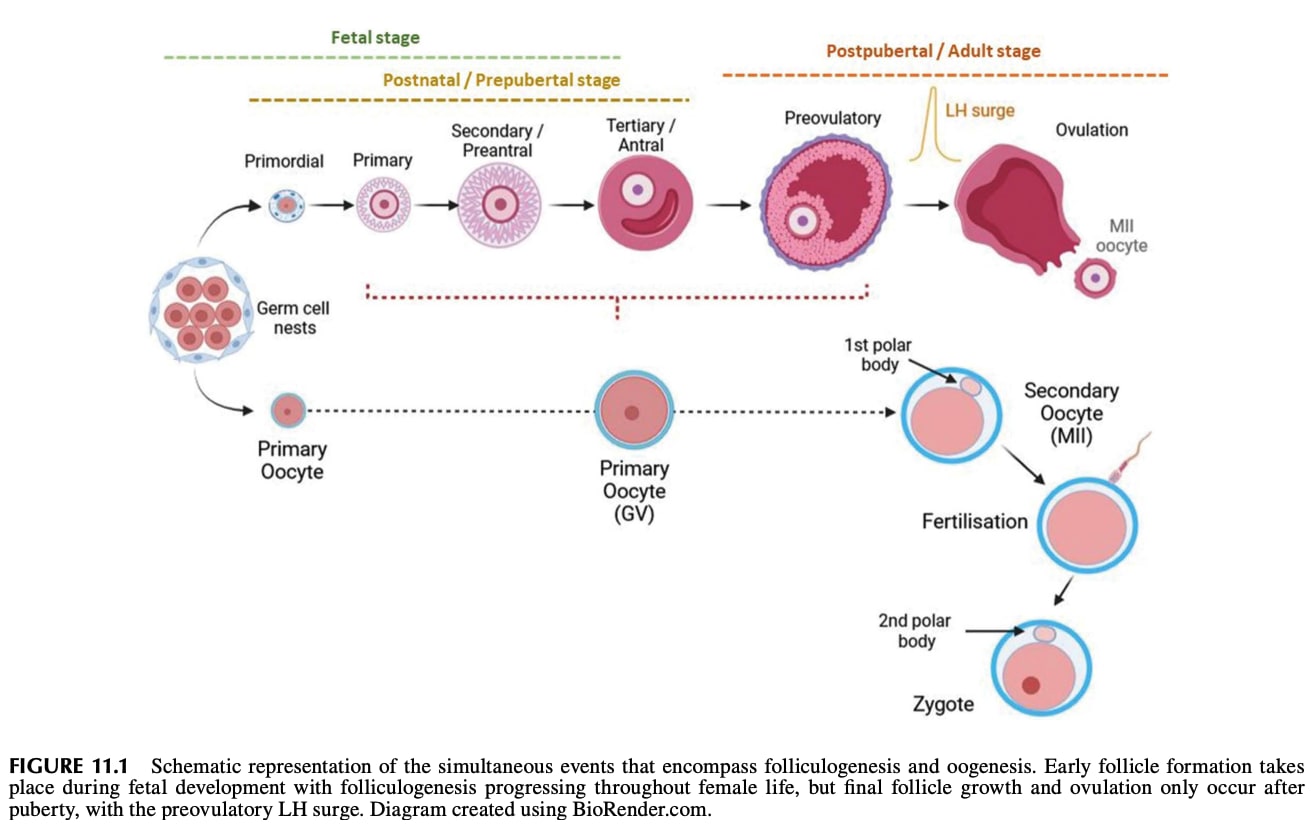
More zoomed in to oogenesis:
[(Figure 11.1 from chapter 11 of Campell and Maalouf 2024[31].)]
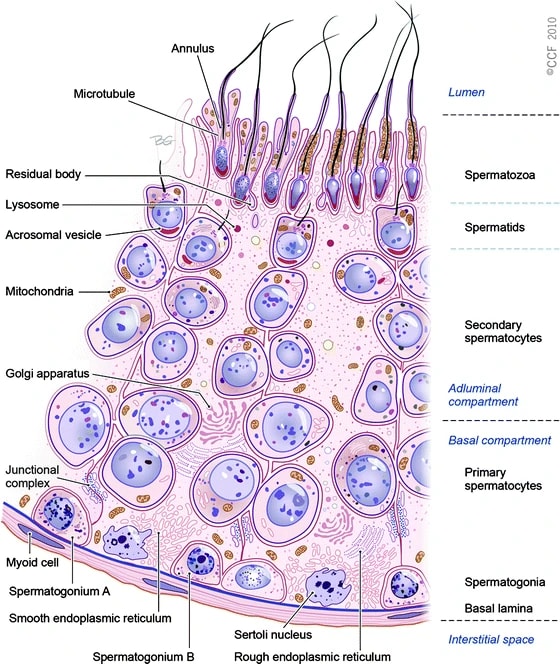
A depiction of spermatogenesis (the cells move from the bottom to the top):
(Figure 2.3 from Sharma and Agarwal (2011)[32].)
IVG is a higher bar than epigenomic correction
A fully successful IVG method takes a stem cell, which could have been genomically vectored beforehand, and creates from it a competent gamete. You could take a somatic cell from an adult, apply your IVG method to make, say, an egg, and then fertilize that egg and make a healthy baby. This is a high bar, and has to fully complete the above three elements of gametogenesis: epigenomic reprogramming, meiosis, and cell development.
To qualify as an EC-making (epigenomic correction) method, an IVG protocol just has to make an epigenomically competent cell. The cell has to have nuclear DNA that has the right on-DNA sex-linked imprints and genome-wide reprogramming. Meiosis and cell development, while important, are not strictly necessary for EC-making.
The three basic elements of gametogenesis are necessary for fully competent oocytes. In natural oogenesis, oocytes grow quite large (>10x larger than progenitor oogonia) with extranuclear cytoplasm and accumulate a lot of cytoplasmic material—mitochondria and ribosomes, metabolic and regulatory proteins, and a large and diverse set of mRNAs. The size and contents of the cytoplasm kickstart and support the development of the very early embryo while the zygote DNA is still largely silenced. For example, in very early embryonic growth, stored ribosomes produce proteins by translating stored mRNAs[33][34]. Meiosis is necessary so that the oocyte genome is haploid.
Only epigenomic correctness is strictly necessary for maternal EC-making. For many reproductive genomic vectoring purposes, it would minimally suffice to use donor eggs. The donor egg could come from a woman trying to have a child via reproductive GV, for herself, or it could come from a woman not otherwise involved in having the child. You take the nucleus (which contains the nuclear DNA of the donor) out of the donor egg, and you inject into the egg a nucleus containing DNA that's competent as maternal nuclear DNA—i.e., it is epigenomically correct. The donor egg provides the size and cytoplasmic material needed for the early embryo to grow. (In theory, one could even use a diploid cell with maternal imprinting, e.g. by using chromosome extraction to make a haploid maternally-EC cell from the diploid.)
In fully natural reproduction, all the elements of spermatogenesis are necessary. Natural spermatozoa (mature sperm) have, besides their DNA, several structures that are important for natural fertilization. A natural spermatozoon has, for example, mitochondria and a tail for swimming; an acrosome to break through the egg's outer barrier; and a centrosome, which helps organize the DNA for the zygote's first division.
But, like with the maternal germline, only epigenomic correctness is necessary for paternal EC-making. Intracytoplasmic sperm injection (ICSI), where you inject a sperm cell directly into the egg, seems to work ok. The injected cell doesn't need a tail (no swimming involved), and skips the acrosome reaction: the egg's outer barrier doesn't have to be broken down, since you're smuggling the sperm inside manually. The centrosome may be necessary, and the egg may have to be activated somehow (there are methods to do so, though they may be inefficient). Further, although oocytes contribute the lion's share of cytoplasmic material needed for early development, sperm may also contribute some important RNAs[35]. But sperm can if necessary be enucleated[36]. Since, unlike oocytes, natural healthy sperm are easy and inexpensive to obtain, there's no issue with using donor elements from sperm.
That said, even as a narrow EC-making method, the approach of IVG is to approximately recapitulate the natural process of oogenesis or spermatogenesis. Through this lens, the idea is that we are using nature's evolved gene regulatory mechanisms to make the maternal or paternal on-DNA epigenetic imprints, and the genome-wide reprogramming. So even if cell development and meiosis aren't strictly necessary for EC-making, the straightforward approach to IVG as an EC-making method is to recapitulate all or almost all of natural gametogenesis.
(In any case, a full IVO method that produces competent oocytes would be great. Besides being a breakthrough treatment for female infertility and for male-male couples, IVO would provide abundant and eventually inexpensive eggs. On its own, abundant eggs would perhaps double the effects of the existing GV method: simple embryo selection. Abundant eggs also makes any subsequent reproductive GV procedures less expensive and more powerful (more chances to try the procedure). Likewise, full IVS would inexpensively give fertility to infertile men and to female-female couples.)
Other general remarks about IVG
-
IVG should be possible.
- We know that the cell processes 1, 2, and 3, described above, are within a pluripotent stem cell's capabilities, because those processes operate in natural gametogenesis.
- Activating these processes should be possible: as long as you replicate the natural signaling environment, support environment, and initial cell state well enough, the stem cell should behave just like it would in vivo. (There may also be gene regulatory shortcuts that jump straight to more developed cell states, skipping intermediate cell states.) If the process mimics natural gametogenesis well enough, the resulting haploid cell will be able to play the role of a natural gamete.
-
Academic IVG research generally says that it is motivated by understanding and potentially improving reproductive health, which is a different motivation from reproductive genomic vectoring.
- Presumably researchers would be quite happy if they could actually generate human gametes competent to make babies, in order to have a general solution to infertility. But a lot of research is aimed at having any model at all, in vitro, of various steps in gametogenesis. Even a slow, expensive, inefficient model can be useful for studying gametogenesis, e.g. to understand the cellular mechanisms involved and maybe to treat specific infertility problems with simple chemical treatments. While this is an admirable goal, and has been until recently the sole source of progress toward eventually having a more practical system for IVG, it's a somewhat different orientation to research.
- The goal relevant to germline engineering is more like: How can we recapitulate, in vitro, the epigenomic reprogramming that occurs in natural human gametogenesis—or mimic the final results—to such a high standard that we can actually use the resulting DNA to make babies? Furthermore, how can we do so reliably and efficiently at scale? Separately, how can we perform meiosis, as is done in natural gametogenesis, but maybe by a different method? How can we accelerate these processes so they don't take many months? How can we make all this happen ASAP, rather than eventually?
- These questions induce different research priorities. For example, IVG research aimed at solving fertility often assumes that, in the context of applications, naturally epigenomically corrected DNA is not available. But for reproductive GV, that assumption doesn't have to be made, so we can consider using naturally EC DNA, or piggybacking off of natural EC-making, or using a prospective parent's own healthy gonadal tissue in vitro.
-
There's no clear line where a method qualifies as in vitro gametogenesis.
- Besides performance (efficiency, cost), different methods have different inputs:
- some kind of stem cell to be turned into oocytes (iPSCs, ESCs, PGCs, oogonia, oocytes, prospermatogonia, SSCs, spermatocytes);
- some kind of gonad-like cells (tissue extract, organoids, reconstituted tissue, xeno gonads);
- and some culture media.
- Different methods also have different outputs: spermatogonia or spermatocytes or spermatids, or immature oocytes or mature oocytes, of varying qualities.
- Because of the *scopes problem in biology, the meaning and measurement of germline cell quality is fuzzy. The highest standard would be to take a somatic cell, and then with no additional cells taken from any organism, produce a cell that is totaly indistinguishable from a natural mature oocyte or a natural spermatozoon.
- Besides performance (efficiency, cost), different methods have different inputs:
-
It is possible to get an IVG-like method that creates cells that are haploid, but that have only some of the needed imprinting.
- So, even if we get a partially working IVG method, some understanding of epigenomic correctness is still necessary. In order to avoid an epigenomic near-miss, we'd need to know when our cell is in a good enough state to be a gamete or zygote. We can't just trust that, since something like gametogenesis happened, the result is a fully competent gamete.
- In any case, a method for inducing meiosis, regardless of epigenetics, would be useful for iterated meiotic selection or recombinant chromosome selection.
The general state of IVG research
We have to rewind a bit before talking about gametogenesis proper. Here's the beautiful illustration we saw earlier, showing the process of natural mammalian germline development:
[(Figure 1 from Saitou and Hayashi (2021)[28:2].)]
Here's a more schematic diagram, with terminology:
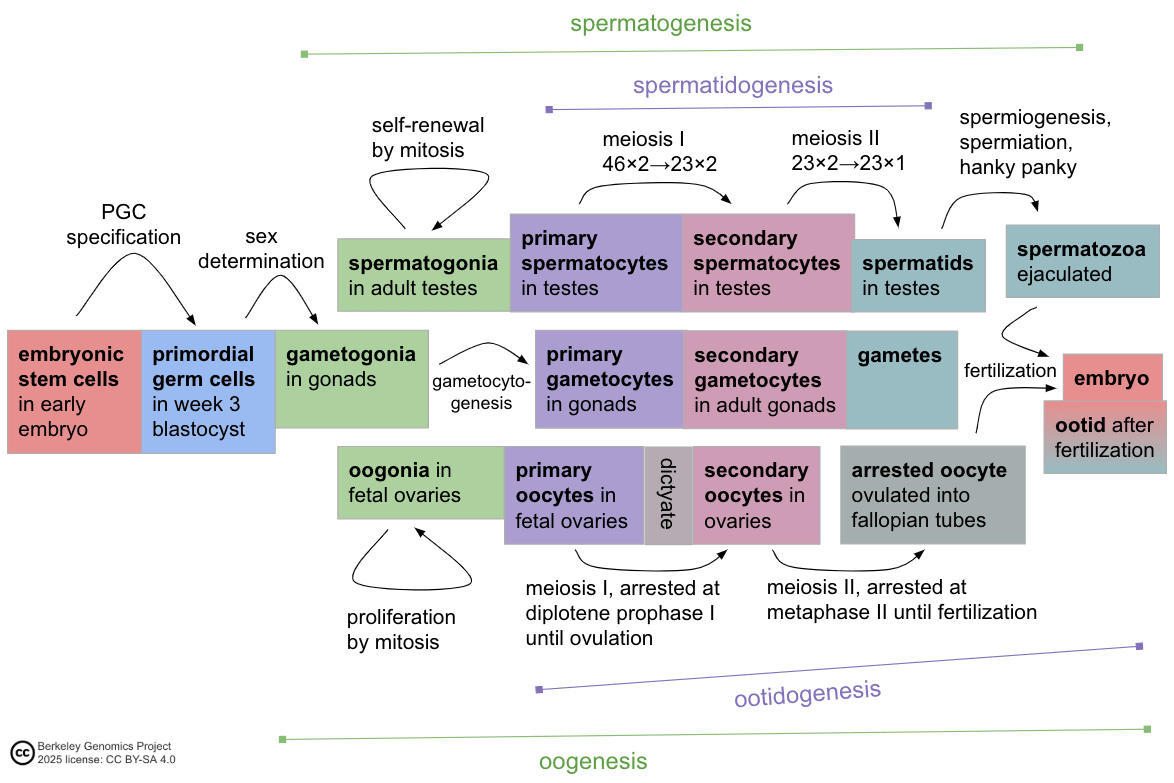
Gametogenesis (oogenesis, spermatogenesis) is shown between the faint green brackets.
Here's a summary of the state of IVG research from 2021:
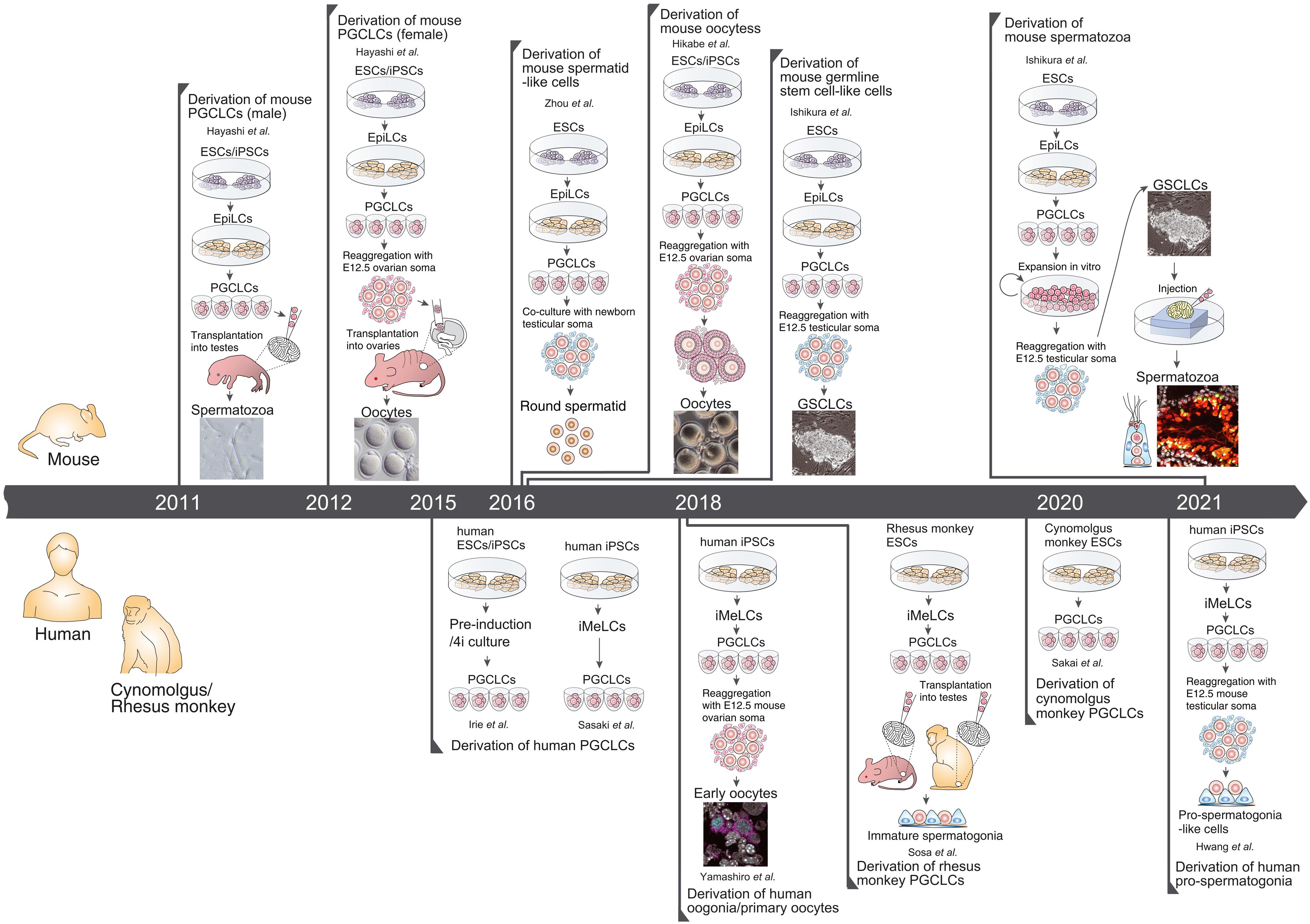
[(Figure 4 from Saitou and Hayashi (2021)[28:3].)]
Most of the progress has relied on culturing stem cells with some sort of special stem cells for support, such as embryonic stem cells or specifically gonadal stem cells, taken from mice or monkeys or humans. Chemical additives (cytokines, etc.) are important, but the support cells are as yet indispensable for the hardest steps of gametogenesis. This suggests that the likely way forward for human IVG will involve ovarian follicle culture, so that area ought to be afforded additional talented researchers; see the "Key challenges" section of Saitou and Hayashi (2021)[37].
Gametogonia are the stem cells that proliferate themselves by mitotically dividing, and that can also differentiate into gametes. Before gametogonia specialize to be female (oogonia) or male (spermatogonia), they have a shared developmental history as primordial germ cells (PGCs). PGCs form in the early embryo, then migrate to the developing gonads, and then differentiate into gametogonia. An illustration of the whole human germline cycle:
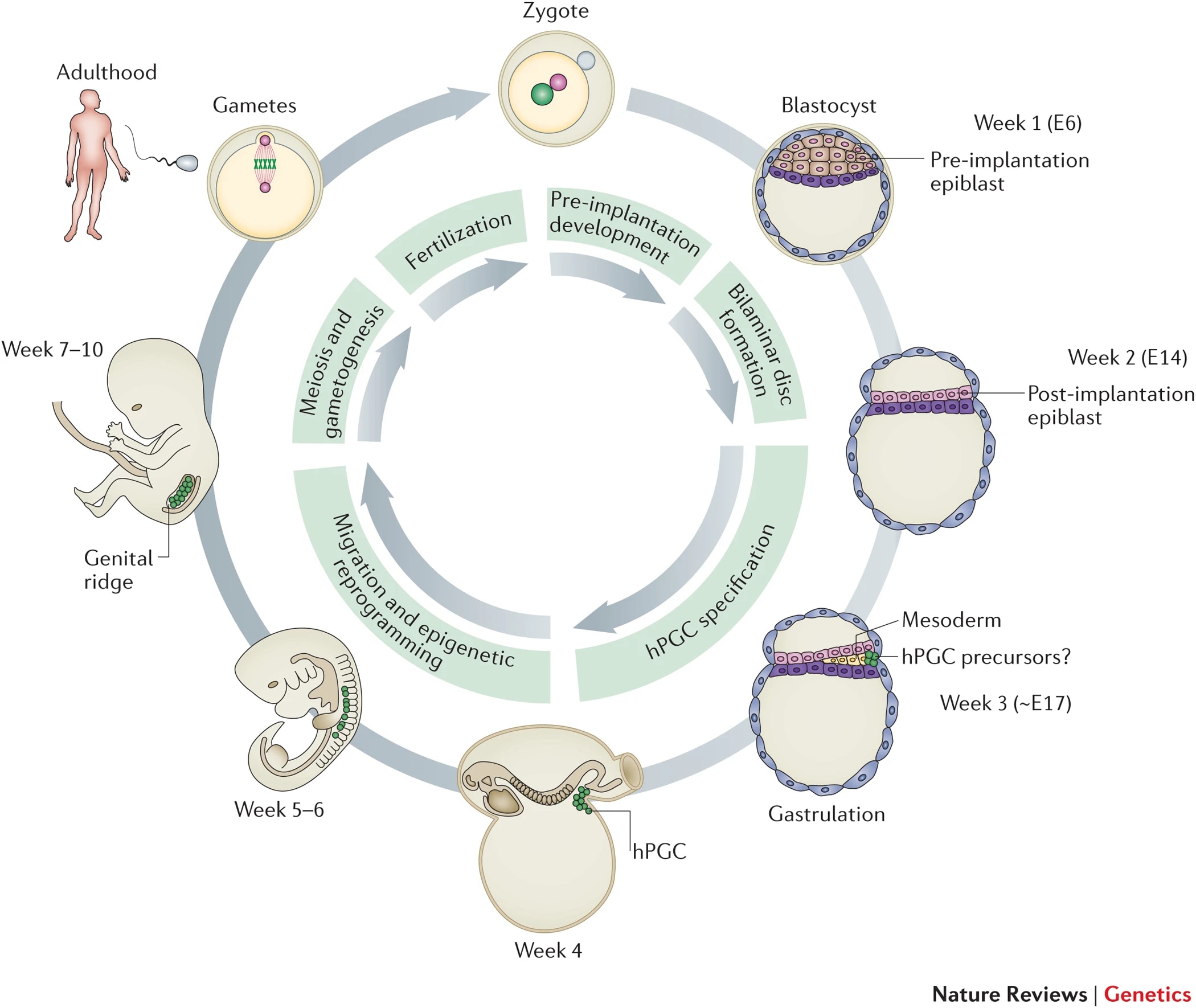
[(Figure 1 from Tang et al., 2016[38].)]
PGCs (primordial germ cells) have a distinct epigenomic state from other embryonic stem cells (ESCs). During germline specification, the ESCs that become PGCs have their epigenetic markers almost completely wiped, including sex-linked imprints, whereas other ESCs mostly keep their imprints as they differentiate to form other fetal tissues. Can we recapitulate this in vitro? Human PGC-like cells spontaneously form, with some low frequency, just from an in vitro culture of human ESCs[39].
We could use ESCs. But if possible, to avoid having to take cells from an embryo, it's better to use iPSCs. Induced pluripotent stem cells (iPSCs) come from somatic (non-stem) cells, taken from an organism's body. These somatic cells are already differentiated, and can't easily be converted into other cells, and generally can't even mitotically divide. Takahashi and Yamanaka (2006) discovered how to use a cocktail of four transcription factors (now called Yamanaka factors) to de-differentiate somatic cells into pluripotent stem cells, which can divide and can potentially differentiate into many kinds of cells[40].
Irie et al. (2015) (using their 4i medium with four inhibitory cytokines), as well as Sasaki et al. (2015), found fairly efficient methods to make human PGC-like cells (PGCLCs) from human ESCs or from human iPSCs [41][42]. (See also Panula et al. (2011)[43].) Together, iPSC induction and iPGCLC induction wipe much of the epigenetic marks from the cell's DNA. This partially mimics the effect of natural PGC specification, and puts the cell in a very undifferentiated state, ready to be reprogrammed into gametes.
Once we're at the PGC (or PGC-like cell) stage, the next step is sex specification, which turns PGCs into gametogonia (oogonia, spermatogonia). The oogonia and spermatogonia will differentiate into primary gametocytes (respectively oocytes and spermatocytes). It's around this time that sex-specific imprinting is established in these germline cells. Finally, the gametocytes undergo meiosis, producing (eventually) gametes. Since these steps are sex-specific, they'll be addressed in their respective following subsections on IVO and IVS.
Here's an illustration of gametogenesis, including epigenomic reprogramming:
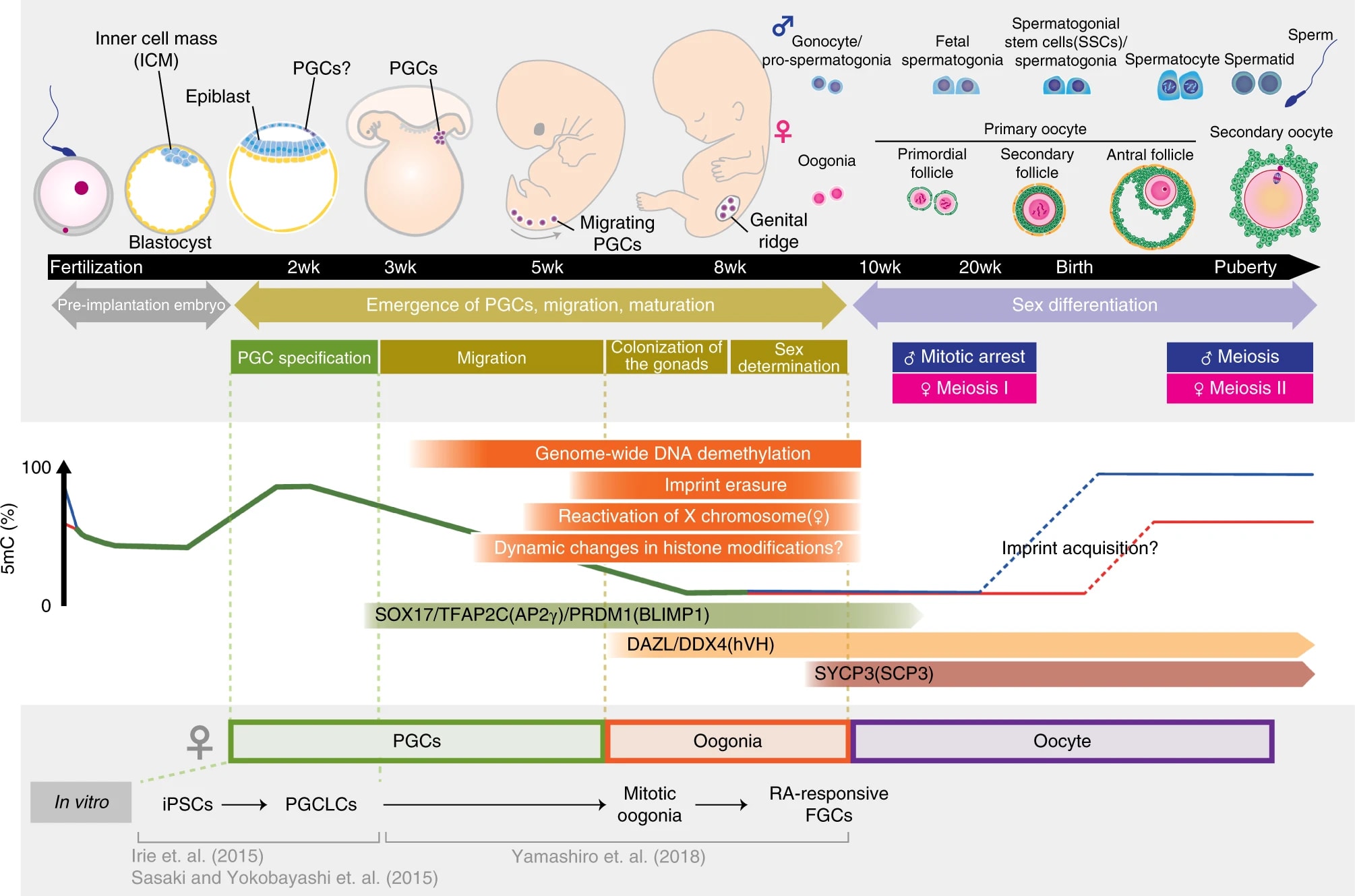
[(Figure 1 from Yamashiro et al. 2020[44].)]
Of special importance for our EC-making purposes is the developmental periods when epigenetic imprinting is established (middle right). The dynamics of imprinting in male and female germlines aren't fully understood, but broadly the picture seems to be:
- Paternal imprinting is established starting in prospermatogonia as they differentiate to spermatogonial stem cells, and is at least largely complete by the time the primary spermatocyte, in adulthood, is partway into meiosis I (pachytene substage of prophase I)[45][5:1].
- Maternal imprinting is established postnatally, in the slow prepubertal growth of the dictyate oocyte[20:1].
A schematic diagram showing when imprinting is believed to be established:
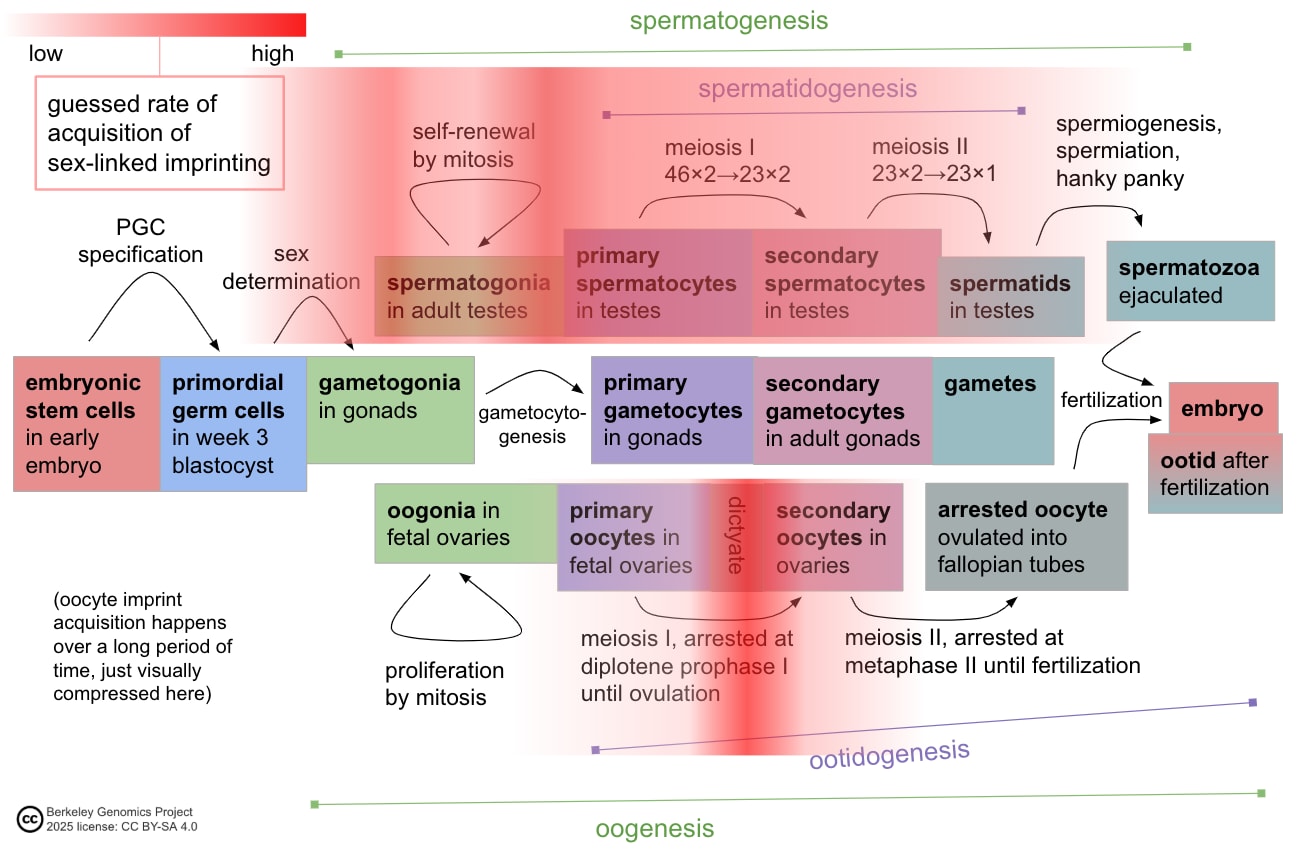
Both IVO and IVS will have to make the cell undergo meiosis. In meiosis, the diploid cell with 46 chromosomes copies its DNA (), performs chromosome recombination, and then divides twice () to produce four haploid daughter cells with 23 chromosomes each. Meiosis is a complicated process that takes at least a few weeks in human cells (generally a few months in natural gametogenesis, depending how you count):
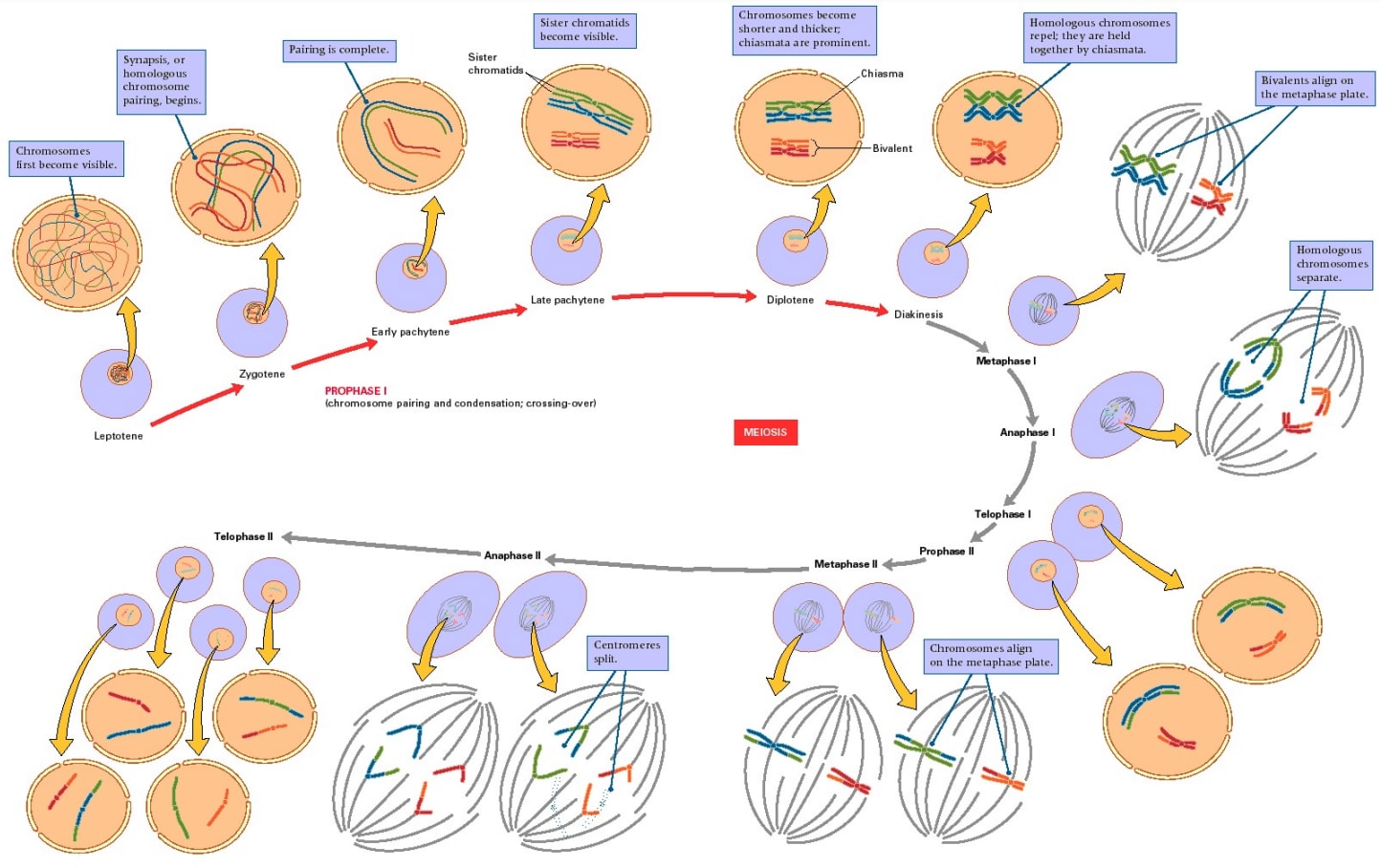
[(I'm unsure where this diagram is from originally; there are several versions floating around. Keser (2016)[46] cites it to Usserly (1998)[47].)]
Ivy Natal is a commercial company aiming to achieve meiosis first in animals for agricultural purposes.
In fact, meiosis may happen spontaneously in cultured human stem cells; several projects have claimed to achieve meiosis in human cells, with only a few or no genetic tweaks[48] (see [27:1] for discussion). However, the efficiency of those methods is quite low, producing <2% haploid cells, and those studies might not replicate straightforwardly or at all. Further, it's unclear what kind of cells they made; some of them may have been dead or dying cells with reduced DNA content, rather than actual haploid cells produced by meiosis. Smela et al. (2024) demonstrate directly inducing human iPSCs to quickly (two weeks) get partway through prophase I of meiosis (maybe 0.1% up to the pachytene substage)[49]. Their method induced >15% of the starting stem cells to enter meiosis:
[(Figure 5G from Smela et al. (2024).)]
A small fraction of the cells expressed MSH4 (the greenish ones in the lower right), a protein that's involved specifically in the pachytene substage of prophase I, suggesting some cells reached that point:
[(Figure 5C from Smela et al. (2024).)]
Further, by culturing fetal testis tissue, Yuan et al. (2020) produced haploid cells and more thoroughly verified their haploidy[50]. In principle, this provides a method for meiosis in vitro, maybe combined with Hwang et al.'s (2020) method for inducing human prospermatogonia-like cells in vitro[51]. This would be an inefficient and non-scalable method (requires human tissue) for inducing meiosis, but shows that it's technically possible in some sense.

As will be mentioned below, both IVO and IVS have already been achieved in mice, in some fashion. But mouse reproductive epigenetics is different from human reproductive epigenetics, so results from mice don't necessarily translate to humans (or other mammals)[38:1]. Part of the reason that mice are a model organism for reproduction is that certain lab mouse cell lines are uniquely easy to coax into certain cellular processes. Also, there hasn't yet been an extensive study of the health of mice grown from eggs and sperm derived via IVO and IVS. Such a study would greatly increase the flow of valuable feedback about which epigenetic imprints are and are not important for completely healthy development, and which of those imprints are and are not corrected by IVG methods.
In vitro oogenesis (IVO)
In vitro oogenesis (IVO) means taking human stem cells and inducing them to differentiate into functionally normal oocytes ready to be fertilized with a sperm and thus contribute to a healthy baby.
Takeaways:
- IVO research has been making progress on oogenesis in the initial stages (up to primary oocytes) and final stages (final oocyte maturation). But there is a core uncrossed gap: maturing a small primary oocyte and primordial follicle from the perinatal stage to the adult stage, where it should be a larger germinal vesicle oocyte in a preantral / antral follicle. This step is crucial because—besides making the oocyte competent on its own to support the growth of the zygote—this step is where maternal epigenetic imprinting is established.
- The most successful methods for making primary oocytes in vitro use human fetal tissue, and therefore are not scalable. There is some progress on methods that instead use only iPSCs, culture media, and targeted gene overexpression.
- More data (scRNA, scATAC, scChIP, bisulfite seq) from natural oogenesis would tell us more about how to control gene expression to activate oogenesis.
- Better methods for culturing ovarian tissue or ovarian organoids, especially fetal and postnatal ovary-like tissue, might help us cross the maternal EC-making gap.
Various research groups are working on elements of IVO. Commercial ventures aiming to treat infertility include Ovelle, Conception, Ivy Natal, Vitra, Gameto, and Dioseve. See the citations in this section for academic groups.
Sneak peek (but note that some of these steps still can't be said to be "solved"):
Morohaku et al. (2016) were able to culture fetal mouse ovaries in vitro ("ex vivo") so that the PGCs in the ovaries grew into oocytes[52]; this method uses whole tissues taken from mouse fetuses. That same year, Hikabe et al. (2016) achieved a kind of end-to-end IVO in mice. They took mouse PGC-like cells created in vitro from ESCs, and grew them in culture with cells taken from female mouse embryo ovaries and with certain culture media. Some of the PGC-like cells developed into oocytes, and when fertilized, those oocytes produced apparently healthy offspring who were themselves naturally fertile[53]. An illustration of their method:
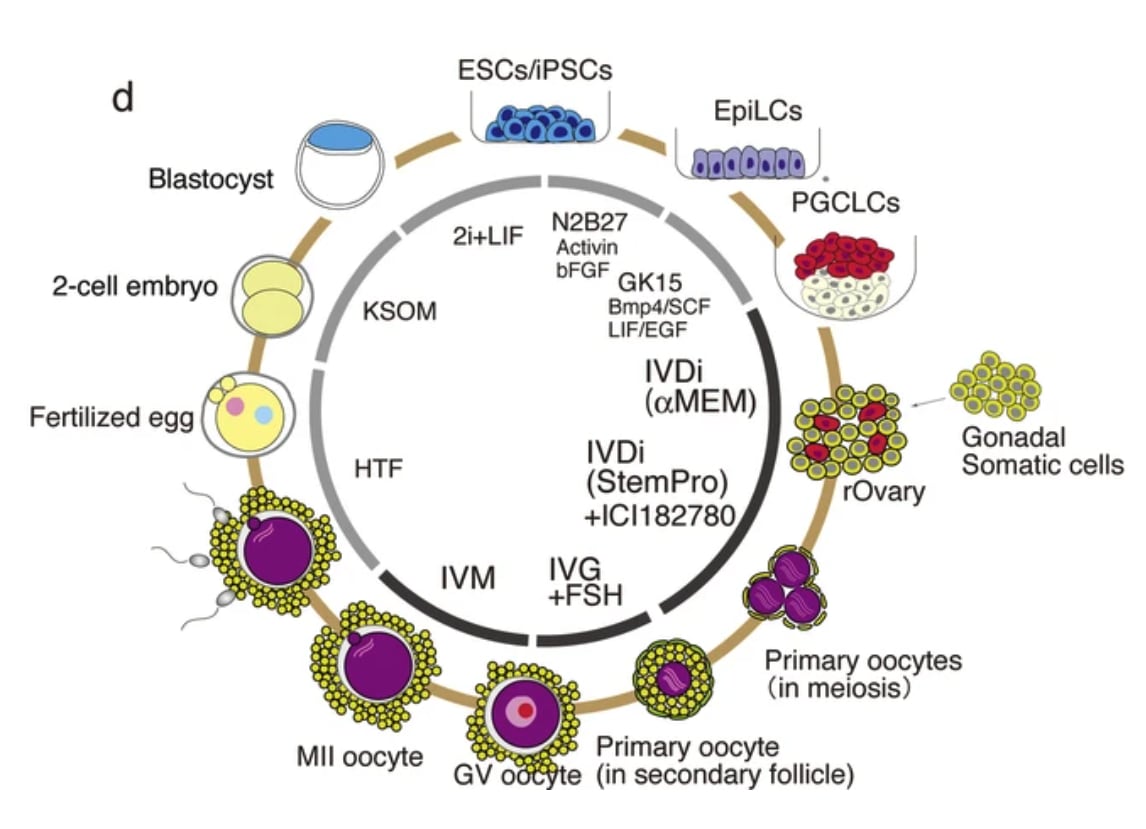
[(Extended Data Figure 9d from Hikabe et al. (2016).)]
The human analog of this method would not be very practical for use in reproductive GV, and mouse reproductive epigenetics is different from human reproductive epigenetics, but it does demonstrate the possibility of mammalian IVO.
When it comes to humans, there's been significant progress on several legs of the female germline's developmental journey, though full IVO has not been achieved. Recall the process of natural oogenesis, depicted running along the bottom half of this diagram:
As mentioned above, we can create pluripotent stem cells from human somatic cells (iPSCs)[40:1], and from iPSCs we can create human PGC-like cells[41:1][42:1]. The next step is to take PGC-like cells and turn them into oogonia, the stem cells that proliferate in the fetal ovaries and then differentiate into oocytes (or die).
Yamashiro et al. (2020) produced human oogonia-like cells from human iPSCs. They induced the iPSCs into PGC-like cells, and then cultured the iPGCLCs with mouse fetal ovary cells, producing oogonia-like cells[44:1]. Their method takes around 2.5 months, and around 10% of starting hiPGCLCs make it to an oogonia-like stage. As they suggest, mouse fetal ovarian cells probably won't be a good platform for human oogenesis proper, because they probably can't support the differentiation of oogonia into meiotic oocytes, especially given the close interaction between a natural developing oocyte and its surrounding ovarian support cells.
The next step from oogonia is oogenesis, where the oogonium differentiates into a primary oocyte, which (eventually) goes through meiosis I to become a secondary oocyte, and then meiosis II to become an ovum. An illustration:
[(Diagram from Slagter et al.[54].)]
The first step is to enter meiosis I. In contrast to spermatogenesis, in oogenesis the primary gametocyte, in this case a primary oocyte, does not complete meiosis I at first. See the gap shown in the above diagram between birth and menarche; in that gap, meiosis is paused. Primary oocytes get most of the way through prophase I of meiosis I, and then stop in the diplotene stage. Most of the meiotic action has already happened at that point. The genome has been duplicated. Homologous chromosomes have paired up and have formed chiasmata, which become DNA recombination sites, where DNA is exchanged (or in small amounts copied) between homologous chromosomes. Zooming in to prophase I on the earlier diagram of meiosis:
As mentioned above, Smela et al. (2024) induced human iPSCs to get to the pachytene substage of prophase I, and some cells looked like fully meiotic cells in terms of their RNA expression[49:1]. Their method somewhat bypasses the PGC and oogonium stages (though there was some resemblance); that might make it harder to produce epigenomically correct oocytes, but on the other hand it's cheaper and faster. Further, their method doesn't use extracted tissue, making it much more scalable. Instead, they use culture media and gene overexpression. They knew which genes to try overexpressing by looking at Garcia-Alonso et al. (2022)'s human gonadal tissue dataset of single-cell RNA-seq (which says what RNAs are in a cell, hence what genes are being expressed) and scATAC-seq (which says which areas of a chromosome are packed/unpacked, hence likely to be inactive/active)[55].
Mizuta et al. (2022) developed methods for taking ovarian tissue from cynomolgus macaque fetuses and culturing them. These reconstituted ovaries could make cyno cells to advance to the diplotene substage of prophase I, according to transcriptomics (sequencing mRNAs). (I'm not sure whether they used cyno iPSCs or just monitored the oogonia in their reconstituted ovaries.) They got similar results with human reconstituted ovaries, where the oogonia present in the extracted fetal ovarian tissue developed to what appeared to be the diplotene stage. Since the method uses tissue from aborted fetuses, it can't scale, but it's a step[56]. No one has made oogonia go all the way through meiosis to produce a mature oocyte.
Natural oogenesis is not a process where isolated cells, destined to be eggs, go through development on their own. Rather, oogonia are connected to each other during proliferation. As the oogonia differentiate into oocytes, they recruit other cells from the ovaries to form follicles, which are support structures that grow with and coregulate the oocyte. Follicles are tightly connected to oocytes: the cumulus granulosa cells, which directly border the oocyte, literally form direct cytoplasmic bridges with the oocyte[57]. Follicles are crucial for supplying nutrients to the growing oocytes, and for regulating the meiosis and reprogramming processes that the oocyte undergoes. Here's a diagram depicting human oogenesis and folliculogenesis:
[(Figure 11.1 from chapter 11 of Campell and Maalouf 2024[31:1].)]
More zoomed in:
[(Diagram from Mescher (2025)[58].)]
The natural process of postnatal, preovulatory oogenesis and folliculogenesis is somewhat murky. Primordial follicles are formed during fetal development and stay stored in the ovaries, with the oocytes arrested in the diplotene substage of meiosis I. But more developed follicles, through the preantral and antral stages of growth, are found in postnatal prepubertal ovaries[59]. On the other hand, prepubertal ovaries are not the same as pubertal ovaries, even leaving aside ovulation; follicles in prepubertal ovaries grow at much lower rates in culture compared to pubertal ovaries, suggesting that follicles and oocytes may continue with important development during all of childhood[60]. Unless this was clarified recently, it's not well-understood what development happens in follicles and oocytes during childhood, so we don't know what it would take to recapitulate that development in vitro, and we don't know how necessary it is to do so in order to make reproductively competent oocytes in vitro.
Thus, there's a gap. We can sort of get from a human somatic cell to a human primary oocyte-like cell, albeit inefficiently and expensively, but as yet we haven't proceeded from that point.
What comes after the gap? The next step would be the final steps of oogenesis: maturation of the follicle and oocyte past the antral stage, partway through meiosis II, at which point the secondary oocyte is ready to be fertilized. This maturation normally happens during ovulation. So to recap, there's the gap, where in vitro differentiation should go, taking primordial follicles to secondary or antral follicles and germinal vesicle oocytes; and then there's in vitro maturation, which takes the grown follicles and produces mature oocytes from them. An illustration:
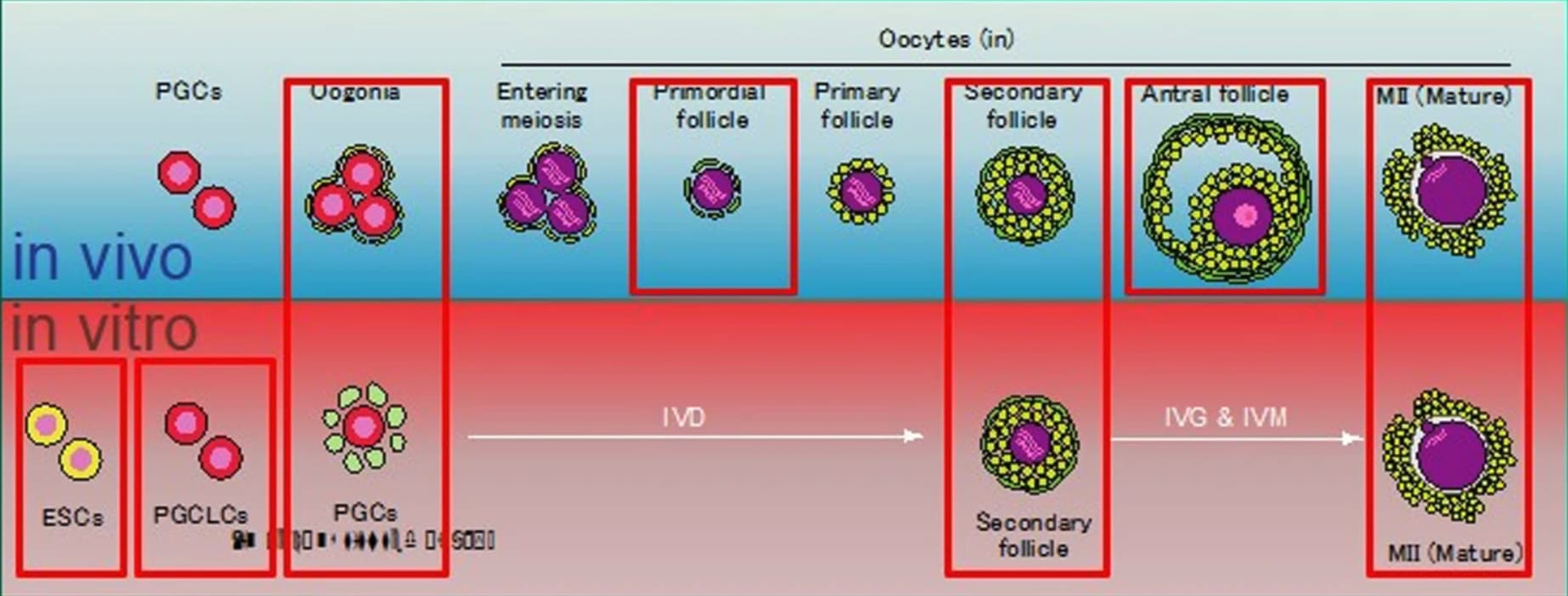
[(Figure 1 from Silber et al. (2024)[61].)]
It's possible to preserve a woman's fertility, e.g. through cancer treatments, by extracting a part of her ovaries. The ovarian tissue contains oocytes in follicles that are in a variety of growth stages. It's then possible to get the oocytes and follicles to mature. They grow and meiose into secondary oocytes ready to be fertilized, producing live human births[62][63][64]. Silber et al. (2024) describe how, basically, squeezing the ovarian tissue produces matured oocytes with high efficiency[61:1].
By stitching all these methods together—creating hiPSCs, then hiPGCLCs, then oogonia-like cells, then dictyate-arrested primary oocyte-like cells with primordial follicles, and then ???, and then in vitro maturation—it looks almost possible to make a (very slow, inefficient, complicated) minimum viable human IVO method:
But in fact, this gap is probably quite significant: It is believed to be when maternal imprinting is established. This is the crucial oocyte growth period, starting with the primary oocyte recruiting a primordial follicle during fetal development, and extending through some amount of postnatal growth, to childhood or adulthood, producing a grown primary oocyte in a secondary or antral follicle[27:2][20:2][5:2]. Natural oocytes also aren't meiotically competent until toward the end of their pre-antral growth phase, though this may be due to the follicle support cells actively suppressing meiosis[65].
To cross this gap, we'd likely have to make some form of artificial ovarian tissue. Possibly this could be ovarian tissue reconstituted from ovarian cells taken from another mammal, but it seems strange for this to work, given how closely granulosa cells interface with the oocyte. Another possibility would be to use human adult ovarian tissue, e.g. from the mother looking to have a child, but this is quite costly, especially since it involves surgery. I also don't expect that it would work; adult ovaries are not the same as postnatal ovaries. More research is needed into growing follicles in vitro[66].
Another approach is to induce stem cells to differentiate into ovarian-like cells, maybe forming 3D organoids. Smela et al. (2023) differentiated human iPSCs into cells that resemble granulosa cells, a key component of follicles[67]. Yoshino et al. (2021) made a functional oocyte by inducing pluripotent stem cells to differentiate into fetal ovarian somatic cell-like cells which could support oogenesis... (say it with me)... in mice[68]:
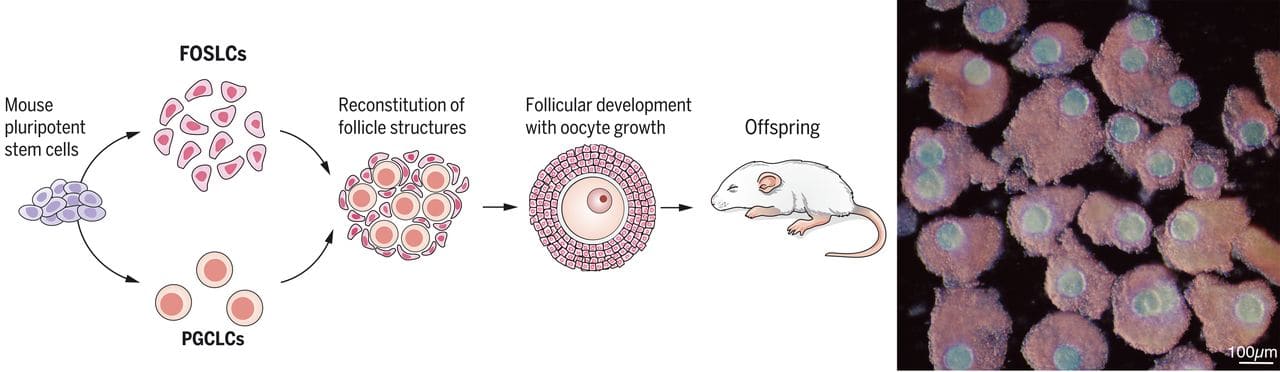
[(Diagram from Yoshino et al. (2021).)]
In vitro spermatogenesis (IVS)
Given more research, it would also be possible to create sperm from stem cells in vitro—i.e., do in vitro spermatogenesis (IVS). This possibility constitutes a path to epigenomic correction that's somewhat independent of IVO; we could plausibly be able to do IVS and not IVO, or vice versa.
Takeaways:
- IVS research, taken together, has already found ways to derive haploid cells from diploid cells through meiosis in vitro.
- Many methods that have been explored are not scalable because they use tissue taken from human fetuses or children. Some methods use adult tissue, which is potentially feasible for adults who are very motivated to use reproductive genomic vectoring, but is probably too costly to support iterative research or GV methods that have to produce many haploids. Methods that use mouse or monkey cells seem promising to explore more.
- Until recently, the results of these studies were not well characterized epigenomically. We haven't had a clear picture of what various stages of the male germline should look like epigenomically, and the cells produced in these experiments haven't been measured epigenomically. This is because single-cell RNA sequencing and large-scale bisulfite methylation sequencing have only recently become widespread, and because single-cell bisulfite sequencing is low-quality. Thus, we don't know exactly how well we're doing, but we can now find out.
Various research groups are working on elements of IVS. One commercial venture aiming to treat male infertility is Paterna. There was another about a decade ago, Kallistem, but they don't seem to be active. See the citations in this section for academic groups.
An illustration of spermatogenesis:
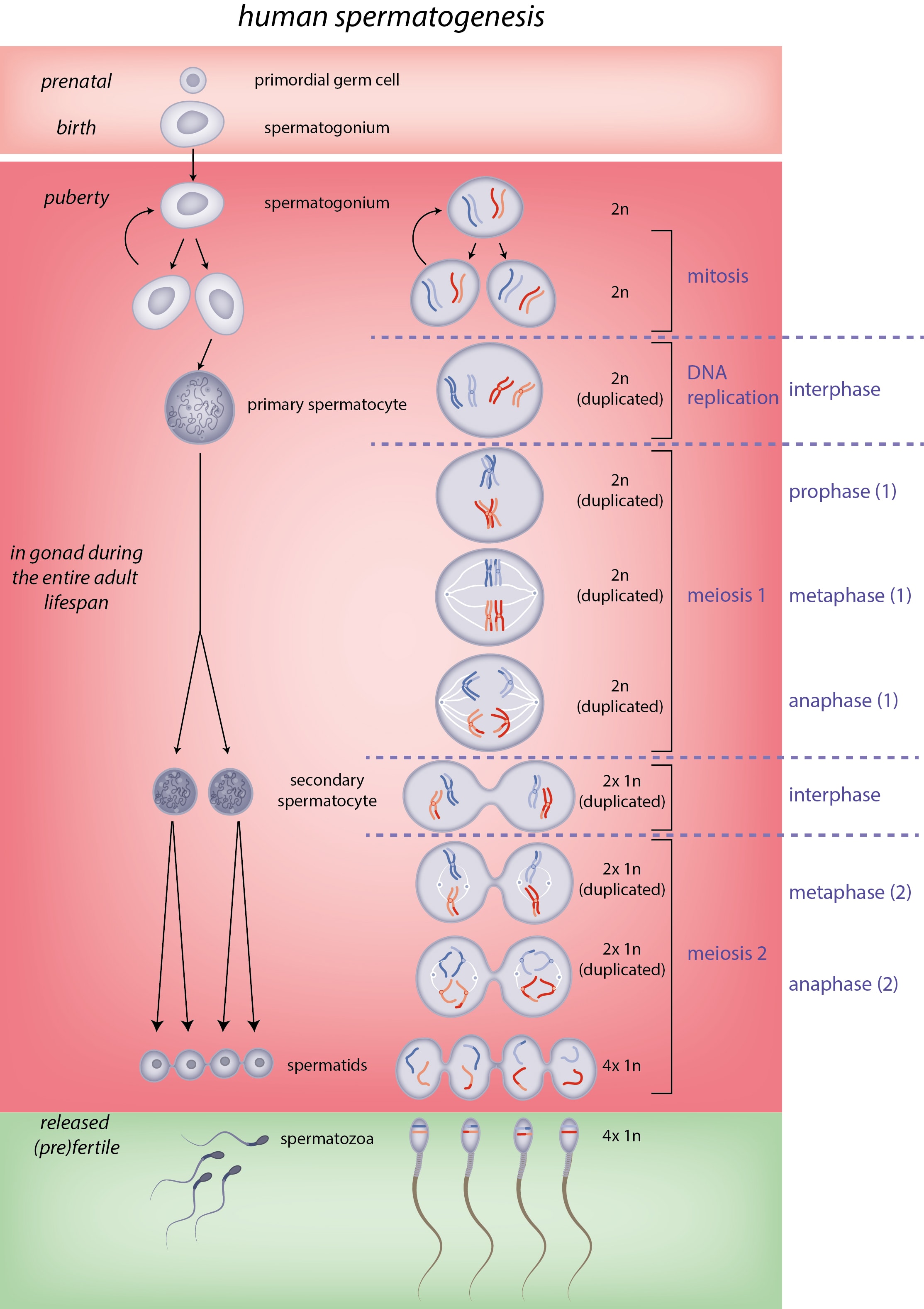
[(Diagram from Slagter et al.[69].)]
Unlike oogonia, spermatogonia are active in the adult gonads, spinning off differentiating spermatogonia which will become spermatocytes. Like oogonia, though, the proliferation and differentiation that happens in spermatocytogenesis involves large groups of differentiating spermatogonia that remain connected to each other. In fact, they stay connected all the way through meiosis (unlike primary oocytes which have separated before meiosis):
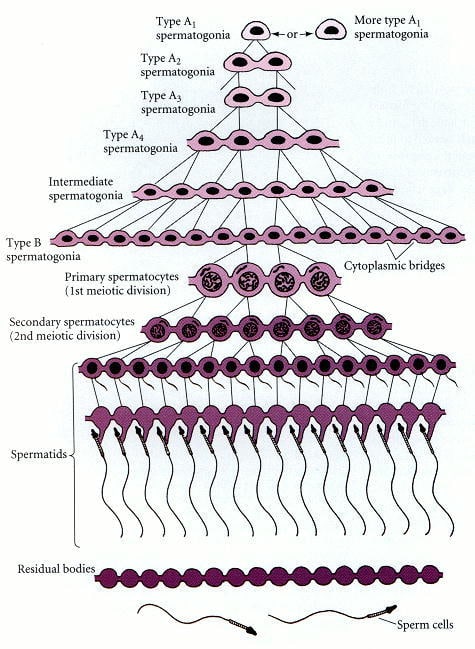
[(Figure 19.18 from Gilbert (2000)[70].)]
IVS has been (minimally) achieved in mice[37:1][71] and rats[72]. Ishikura et al. (2021) reconstituted the whole mouse germline process in vitro[73]. They used reconstituted mouse testes, i.e. blobs of cells from mouse fetal gonadal tissue:
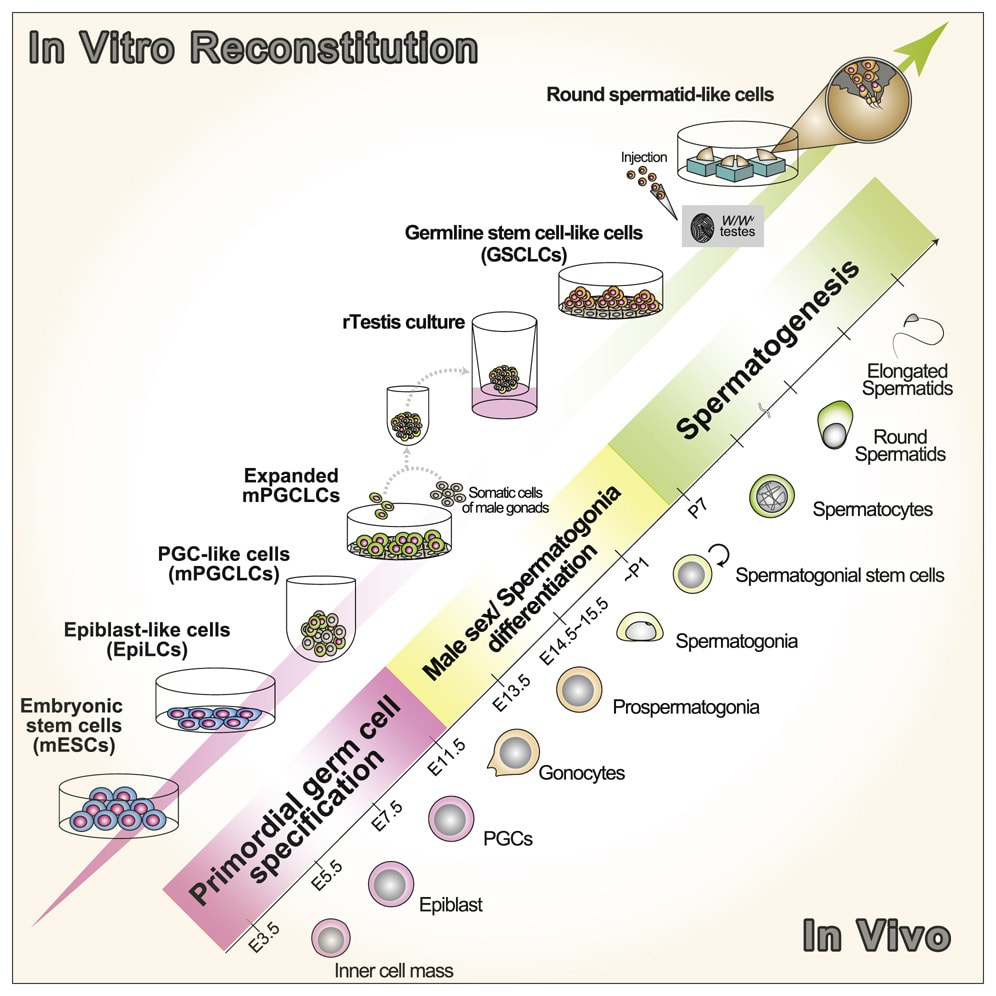
[(Graphical abstract from Ishikura et al. (2021).)]
Recall the diagram of gametogenesis, with spermatogenesis running along the top half:
As we've seen, we can create hiPSCs[40:2] and thence hPGCLCs[41:2][42:2]. The next step is to derive spermatogonia.
Recently, Whelan et al. (2024) induced iPSCs to differentiate into spermatogonia-like cells using a somewhat wilder method: they transplanted mouse fetal testicular cells, along with human PGCLCs, into an adult mouse[74]:
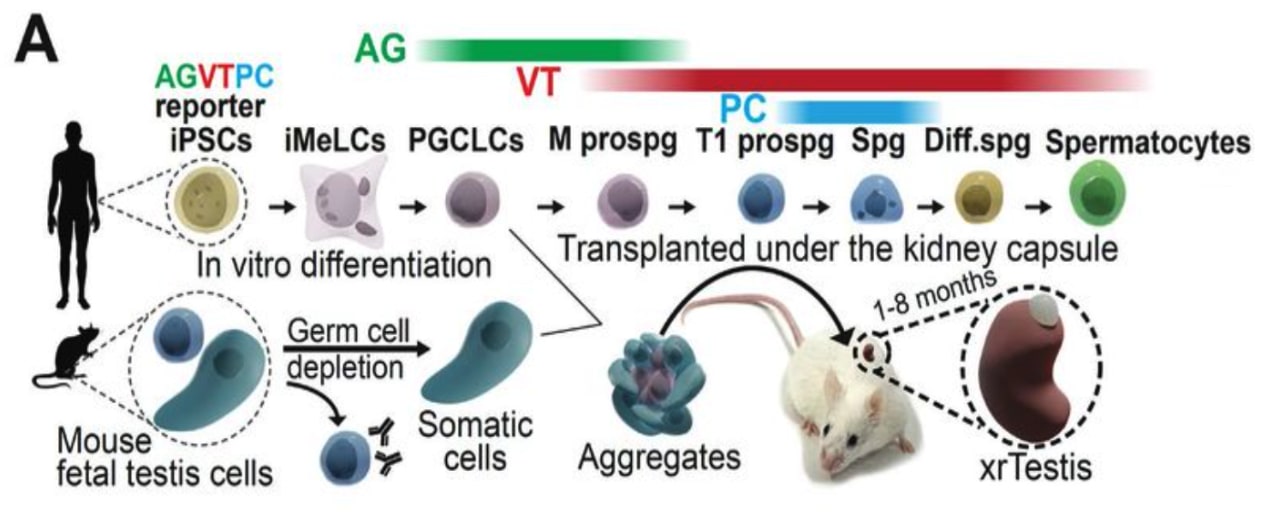
[(Figure 1A from Whelan et al. (2024).)]
The transplanted mouse testicular tissue then recruited blood vessels to feed it, and furthermore regrew 3D tube structure (which I hear is a normal enough result when scientists do this sort of transplant, but still!). Many of the hiPGCLCs differentiated into spermatogonia in the xenografted human-testes-like tissue. Based on scRNA-seq data, a small fraction of the human spermatogonia-like cells appeared to enter the pre-leptotene substage of prophase I, at the very beginning of meiosis I:
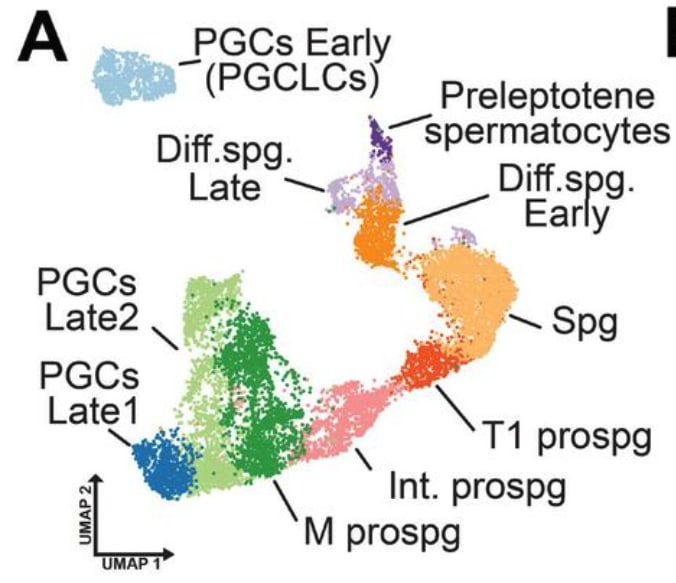
[(Figure 2A from Whelan et al. (2024)[74:1].)]
This method is complicated. Also, it seems that their spermatogonia-like cells may not have started to establish paternal imprints. If that's the case, then at best the cells are more like prospermatogonia. Recall that paternal imprinting likely starts to be established already by the time prospermatogonia are differentiating into spermatogonia.
There have been several studies attempting to recapitulate human spermatogenesis in vitro. Specifically, they're attempting to derive from spermatogonia or primary spermatocytes, in vitro, some haploid cells (round spermatids, elongated spermatids, or spermatozoa) that are competent to contribute to an embryo. See the reviews by Tanaka and Watanabe (2023)[8:2] and Robinson et al. (2023)[29:1]. An illustration of types of IVS methods that have been tried:
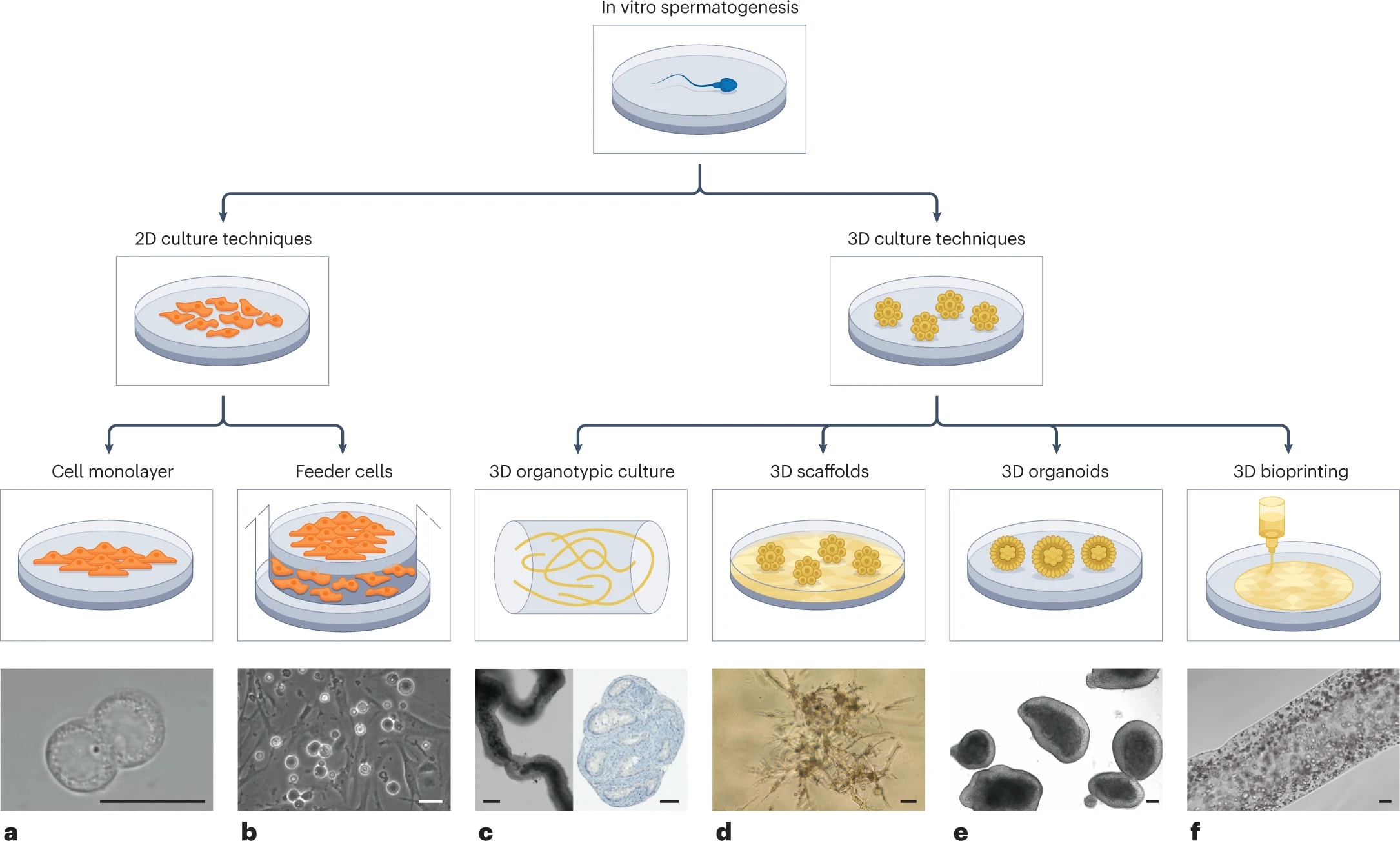
[(Figure 2 from Robinson et al. (2023).)]
I summarize some of these studies in the appendix "In vitro spermatogenesis studies".
Below is a diagram summary of the paper summaries, sketching the state of the art of IVS in humans. Down the middle is the progression of in vitro recapitulated male germline development, through spermatogonia, spermatocytes, and then haploids (spermatids, spermatozoa). The arrows show the transitions achieved in vitro, and are labeled with the study. The fainter the arrow, the worse overall utility of the protocol from the perspective of making usable gametes. Most importantly, epigenomic correction for sperm, especially paternal imprinting, has not been achieved with a high degree of correctness.
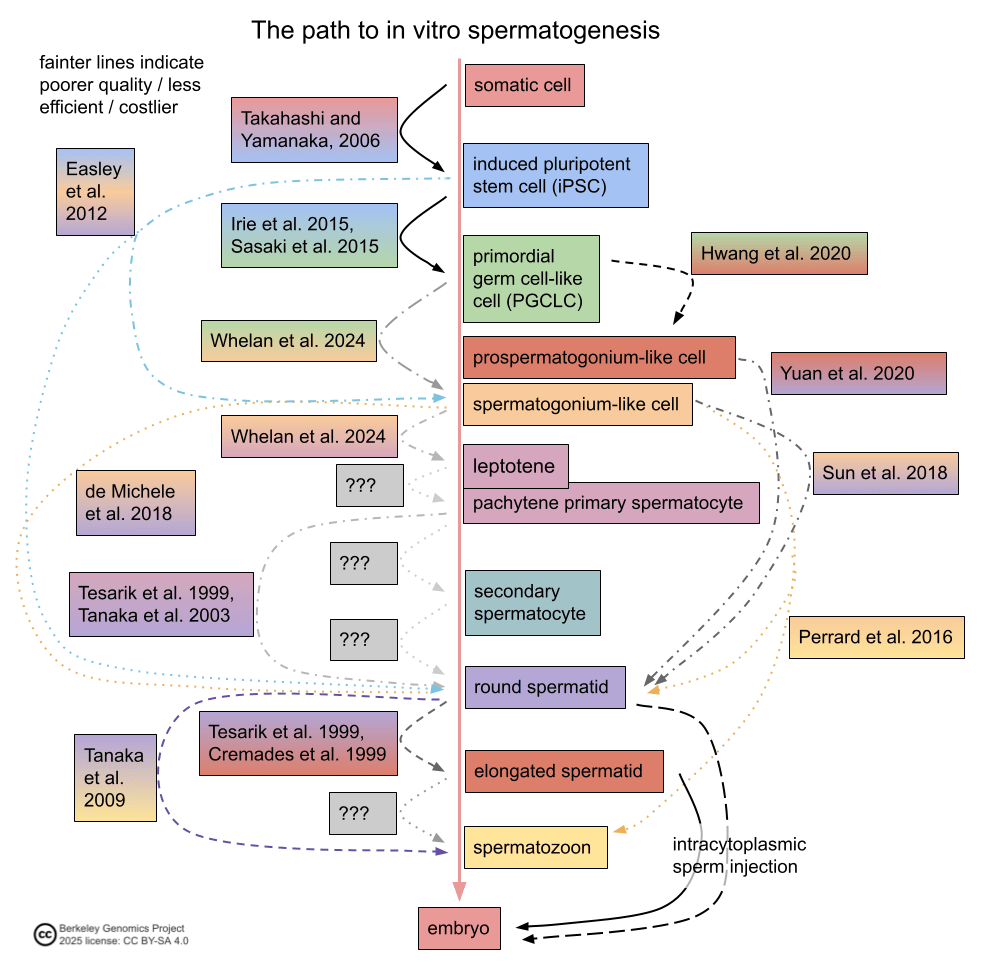
A potential key issue for in vitro spermatogenesis is that testes have a specific 3D structure that supports spermatogenesis. There's a spatial structure to gene expression within testicular tissue[75], and primary spermatocytes cross a boundary called the Sertoli cell barrier before proceeding through meiosis. This means that, compared to IVO, IVS might require a more complicated method to mimic the 3D structure of testis tissue—hence all the IVS experiments with various kinds of culturing methods.
The paternal EC-making gap remains: we haven't been able to make iPSC cells develop into haploids with correct paternal imprinting.
Using natural epigenomic correction
The idea of natural EC-making is to piggyback off of natural processes that convert non-gamete DNA into gamete DNA. Generally, this would work like this:
- Genomically vector some type of stem cell in vitro.
- Transplant the GV cells into gonads or gonad-like tissue.
- Wait.
- Retrieve the resulting gametes.
The point of using natural EC-making is that it could be feasible sooner than artificial EC-making. Generally the reason is that, at least in principle, we don't have to know as much before we can do natural EC-making, compared to how much have to know before we can do artificial EC-making. So we wouldn't have to wait for as many large research projects to succeed. There are two reasons:
- With natural EC-making, we don't have to come up with methods that coax cells through germline development. The natural gonadal tissue already knows how to do that.
- With natural EC-making, we don't necessarily have to know what constitutes an EC state, any more than we have to verify the epigenomic health of every baby who's conceived today by natural means.
No clear boundary between natural and artificial EC-making
These methods are somewhat contiguous with IVG, in that one kind of approach to IVG aims to induce differentiation, meiosis, and maturation in gametogonia by using one or more of:
- in vivo gonads (i.e. a person's ovaries or testicles),
- gonadal tissue (e.g. extracted from adults, or taken from fetuses who died),
- reconstituted gonads (lumping together unstructured cells from gonads in vitro),
- xenogeneic reconstituted gonads (e.g. using mouse ovarian cells to culture human oogonia),
- transplanted/induced in vivo reconstituted gonadal tissue (e.g. growing human testicular tissue inside a mouse by injecting human stem cells or gonadal cells), or
- gonadal organoids (artificially differentiating stem cells into gonadal cells, e.g. granulosa-like cells).
There's no clear boundary between IVG and natural epigenomic correction, and there's no need for one. But there is a fuzzy spectrum between artificial and natural epigenomic correction, that is useful for orienting in terms of "possibly feasible sooner but slow and costly" vs "feasible later but fast and inexpensive":
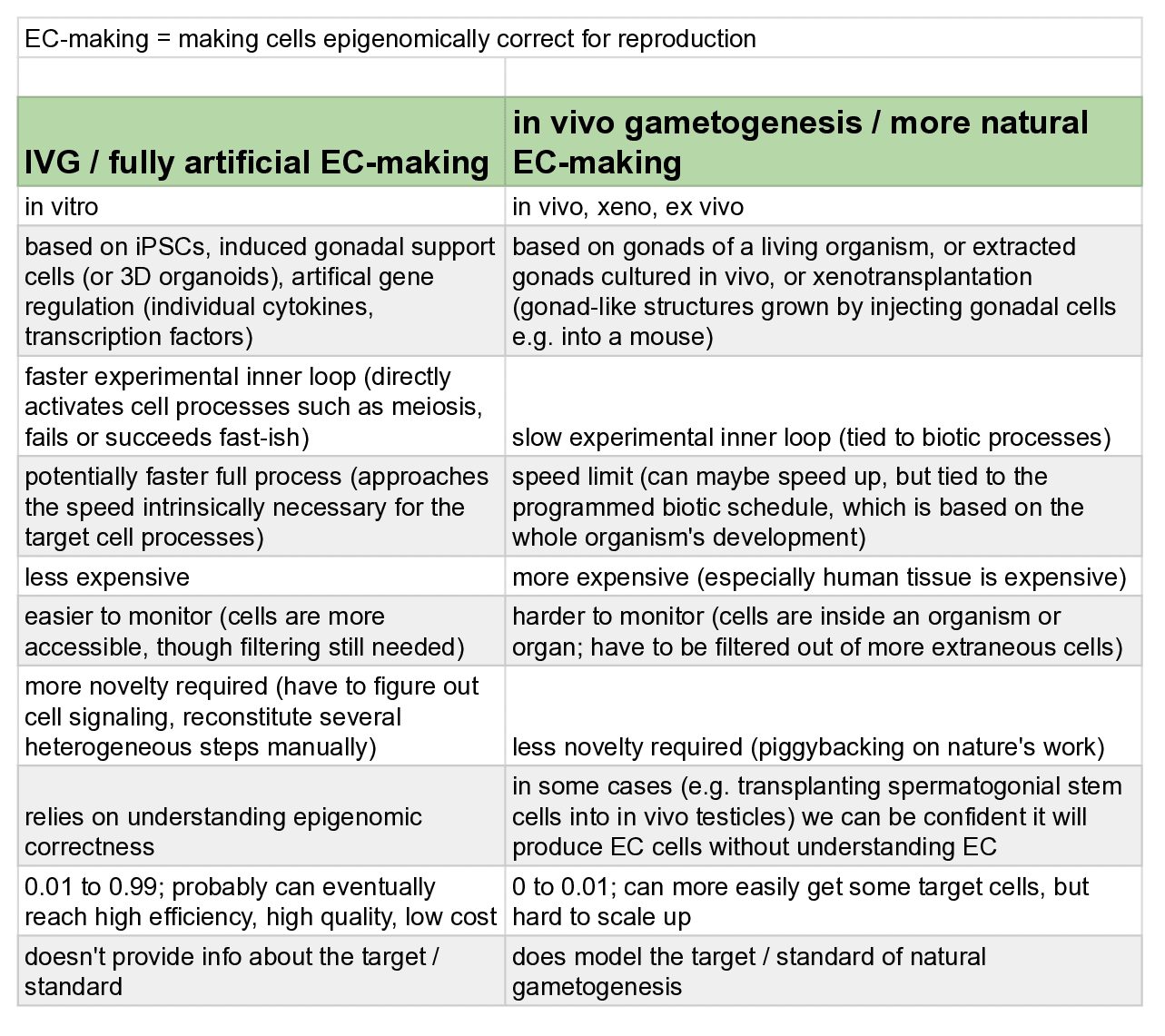
Xenotransplantation (of human gonadal tissue into a non-human animal), for example, is an in-between case. I'm counting ex vivo (culturing natural gonads in vitro after extraction) as being natural EC-making, even though it's not fully natural.
The timing problem with natural EC-making
It sounds nice in theory, but unfortunately there's a big obstacle. Sex-specific epigenetic imprinting seems to be partially established already by the adult stage. In the male germline, adult spermatogonial stem cells seem to already have some of their needed imprinting [45:1][5:3]; and in the female germline, imprinting seems to be established during the slow maturation of oocytes and follicles before puberty[20:3]. Adult gonads are likely to not be able to support the changes in germline cells that perinatal / prepubertal gonads support.
What this means is that the straightforward way of using natural EC-making doesn't work. The straightforward method would be:
- Use some GV method on iPSCs to make GV cells.
- Transplant the GV cells into a living person's gonads (perhaps with additional reprogramming, e.g. induction into a PGCLC, gametogonia-like, or primary oocyte-like state).
- Wait.
- Retrieve the resulting gametes.
But since natural EC-making happens during childhood, this can't be used as an EC-making method because it would involve unnecessary, invasive, dangerous experiments on children.
Alternative gonadal tissue
Are there other ways to get gonadal tissue that can support natural-ish EC-making? Some hypothetical possibilities:
- Adult gonads for full EC-making.
- It is possible that adult gonads might support maturation of immature germline cells.
- In other words, I don't know that it's impossible for oogonia-like cells, transplanted into a woman's ovaries, to mature into germinal vesicle oocytes. Probably not (and someone might know that it's not possible), but maybe. One observation that suggests it might be possible is that ovaries tend to contain oocytes and follicles at range of different growth stages, which weakly suggests that maybe larger steps in oocyte development can be taken in adult ovaries (though it could very well be that in fact all oocytes in adult ovaries already have their imprinting, or the ones that don't cannot gain imprinting in adulthood). If this is possible, it could follow an application of Yamashiro et al. (2020)'s mouse fetal ovary culture method to make oogonia-like cells from hiPSCs[44:2]. There would still be a major issue of retrieving the genomically vectored oocytes—normally only a few hundred are ovulated in a woman's life.
- Likewise, I don't know that it's impossible for prospermatogonia to differentiate into adult spermatogonial stem cells in an adult man's testicles. If so, then it could follow some descendant of Hwang et al. (2020)'s protocol for making prospermatogonia-like cells[51:1]. Some counter-evidence is Sosa et al. (2018)'s experiments. They transplanted rhesus macaque PGCLCs into adult rhesus testicles. They found that the injected cells made some progress differentiating toward prospermatogonia, but lacked at least one marker of prospermatogonia and didn't contribute to spermatogenesis[76].
-
Xeno gonads for full EC-making.
-
A hypothetical possibility is to use non-human perinatal primate gonads, in vivo. In other words: transplant human iPGCLCs or gametogonia-like cells into a baby monkey's gonads. You might have to first kill the germline cells in the monkey's gonads. Then you wait, and later retrieve matured human germline cells. As long as the human germline cells have matured past the point where imprinting has been established, this would suffice as an EC-making method; it could be followed with in vitro maturation of the gametes.
-
This method might take something like a couple years, depending on the species of non-human primate used. E.g. marmosets take around 1.5 years and cynomolgus macaques take 4 or 5 years to sexually mature, which would be the analog of the human imprinting stage.
-
There are still ethical issues with a protocol like this, but they aren't that bad. The monkey could lead a long and healthy life, except for not being able to have genetic children. However, I'm not sure when—prenatally or postnatally—the hPGCLCs would have to be injected. If they'd have to be injected before natural birth, then e.g. a premature caesarean section would hurt the mother and put the baby at risk.
-
Of course, there would be risks that the epigenetic imprinting, and generally the epigenomic state, wouldn't be correct. There's some theoretical reason to think that the imprinting might be ok: probably the imprinting is normally established in detail by intracellular regulatory processes, which only have to be coarsely activated exogenously. In other words, it's likely enough that the support cells surrounding a germline cell are guiding the germline cell through activations of its own machinery, and they don't have to exert detailed control over the germline cell's epigenetic state.
-
That said, this sort of method would have to at least be validated with epigenetic sequencing. So it would require gold standard measurements—epigenetic data about natural human gametes. Thus it can only possibly bypass half of the EC problem (making the right epigenomic state) and can't bypass the other half (knowing what the EC state is).
-
For oogenesis in particular, this method is extra suspect. During oocyte and follicle maturation, oocytes form cytoplasmic bridges with the adjacent cumulus granulosa cells. The granulosa cells support the oocyte with nutrients, and coregulate with the oocyte. It's conceivable that the resulting oocyte would be fine, and we could validate the results at least in terms of on-DNA epigenomics and general RNA and protein content. But it seems at the very least a major cause for concern. In spermatogenesis, by contrast, germline cells are influenced by signals from surrounding gonad cells, and form bridges with each others as they proliferate, but don't form cytoplasmic bridges with non-germline cells.
-
-
Long-term culture of fetal gonads.
- IVG researchers sometimes use gonads extracted from human fetuses that were aborted or miscarried. If it were possible to expand these tissues in vitro and maintain their fetal-gonad-like function indefinitely, they could be scaled up and used for EC.
- This is kinda sci-fi though. I don't know of tissues being expanded and maintained in vitro for multiple years. (Several months, though, is not unheard of, and immortalized stem cell lines are used. And of course tissues live in organisms for years—though not developmentally frozen.)
A more thorough literature search that what I've done is called for. See the reviews already cited[27:3][28:4][8:3][29:2], and other recent reviews such as [77], [78], and [79] (which I haven't read).
Using natural reproductive DNA
The general idea is to use DNA from natural reproductive cells (sperm, egg, zygote, embryonic stem cell). That DNA will be in a correct epigenomic state. One can then apply a GV method that only does operations that don't mess up the epigenomic state, and then use the resulting cell as a gamete or zygote.
These methods are not EC-making methods—they don't correct anything about the epigenome. They solve epigenomic correctness without doing epigenomic correction. One could call them "EC bypass methods".
Examples:
- simple embryo selection;
- gamete selection;
- single-round embryo prime/base editing;
- chromosome selection on spermatozoon DNA, mature oocyte DNA, or ESC DNA;
- iterated CRISPR editing on naive ESCs with imprint maintenance;
- iterated CRISPR editing of SSCs in vivo ("magic rainbow sperm");
- chromosome selection on fully grown germinal vesicle primary oocytes, in theory;
- iterated CRISPR editing or chromosome selection on spermatogonial stem cells, with testis transplantation;
- chromosome selection on immature oocytes, with ovarian transplantation.
Natural EC interrupt methods
The last two methods listed above are border cases, which could be called "EC interrupt" methods. They operate on SSCs or immature oocytes—cells which have already been partially but not fully epigenomically corrected. They use natural, partially-EC reproductive DNA, and they also later apply natural EC-making / piggybacking methods.
Natural EC interrupt methods "pause" natural gametogenesis to do GV, before resuming natural EC where it was paused. They don't do any EC-making of their own.
Chromosome selection on grown primary oocytes is an extra strange border case, where the cells have already received all or almost all of their maternal imprinting, but have not yet meiosed. So technically the natural EC-making has completed, but the broader gametogenesis process, which includes meiosis, has to be continued.
An EC interrupt method requires methods for:
- Obtaining the partially EC cells to apply GV to. E.g. gonadal tissue biopsy.
- Maintaining whatever partial imprints and cell-type niche that the cell already has, while applying GV. See the next subsection.
- Completing epigenomic correction after GVing. E.g. retransplanting the GVed cells.
Imprint maintenance problems
EC bypass methods, which use natural EC DNA, apply GV to a cell that is already completely corrected or, in interrupt methods, partially corrected. The imprints that are already established have to be maintained during GV. The requirements and solutions for imprinting maintenance presumably depend on context (which sort of imprints you have to maintain, and what else you're doing to the cells).
For example, MacCarthy et al. (2024) found a modification of the SOX Yamanaka factor that makes the Yamanaka-like cocktail induce and maintain a naive ESC state[80]. In theory you could edit naive ESCs over several rounds while maintaining the imprinting, and then just use one of the edited cells as an embryo. However, there would be some amount of loss of imprinting due to being cultured and due to the action of super-SOX. (One might be able to then fix the hopefully not-too-numerous aberrations with epigenetic editing.) Further, there's some indication that the rapid reprogramming (e.g. genome-wide demethylation) that occurs in very early embryonic development might already be enough to disrupt an ESC's ability to act as a zygote—e.g. Mitalipov et al. (2002)'s rhesus macaque blastomere SCNT experiments had poor pregnancy rates and one stillbirth (though not necessarily due to developmental abnormality)[18:1]. On the other hand, Meng et al. (1997) also did SCNT to rhesus macaque blastomeres, and out of 29 embryos in 9 females got one 30-day miscarriage and two apparently healthy live births[81]. More study needed.
Broadly speaking, the key question is: How, and how well, can the epigenomic state be maintained during culture in vitro, while applying some genomic vectoring method? If a near-perfect maintenance method is found, further epigenetic correction wouldn't be needed.
Bayerl et al. (2021) found a culture medium that seemed to maintain imprinting in ESCs cultured for several cell passages, though there was still loss of imprinting within a couple weeks (see figure S3H in the supplemental information)[82]. Tan et al. (2020) were able to characterize and culture SSCs as distinct from differentiating spermatogonia, which hadn't been well-separated before[83]. They didn't report on imprinting status of their cultured SSCs, so we don't know their quality. It's plausible that a "close enough" SSC niche would automatically maintain any important imprinting, but it's also plausible that it wouldn't.
It might be fairly feasible to maintain imprinting in culture. There are natural mechanisms that maintain imprinting during very early embryonic demethylation. The paternal genome is very quickly mostly demethylated, even before the first cell division, but the imprints remain (as well as some other methylations, e.g. to suppress transposons). Some proteins (Stella, Zfp57, and Trim28) are known to be involved in protecting paternal imprints[35:1]. Fischer et al. (2025) used Zfp57 to protect imprints somewhat during naive hPSC induction, and they discuss other protective proteins[84]. (Though, they also tried Stella (DPPA3) and found no effect—perhaps Tet3 wasn't around?)
Epigenetic CRISPR editing
CRISPR-Cas9 was a breakthrough in DNA editing in part because, in contrast to previous DNA editors with protein-based binding, it could be fairly easily, flexibly, and precisely targeted to approximately any specific region of DNA using synthesized guide RNAs that bind to complementary DNA single strands. A further innovation was to deactivate the elements of Cas9 that cut DNA, creating dCas9, which doesn't cut DNA. You can attach thingies to dCas9 that have various effects on and around the DNA that the system binds to. Some of those thingies will cause some epigenetic state to change (perhaps by recruiting cellular machinery)—e.g. methylate some CpG islands, chemically modify some histones, or affect chromatin accessibility. Various such systems have been developed in the past decade[85][86].
The idea for EC-making with epigenetic CRISPR editing would be to make a GV stem cell, and then apply epigenetic CRISPR to the cell. This could be haploid or diploid:
- Haploid epigenetic editing: add either maternal imprinting or paternal imprinting, and then use the result as the maternal / paternal DNA contribution to a zygote.
- Diploid epigenetic editing: add both maternal and paternal imprinting to a diploid cell, and then use the result as a zygote or as the DNA contribution to a zygote (via SCNT).
- However, it might be infeasible to usefully edit a diploid genome this way. By default, at least for autosomes, you'd get edits on both homologous chromosomes, so you wouldn't have the appropriate differentially methylated regions or other monoallelic state.
- If there happen to be DNA differences between the homologs, e.g. a nearby SNP, those could be used to target just one allele on a specific chromosome.
- You could calibrate the efficiency of the editor so that on average some fairly small portion of target loci get the epigenetic edit. Then you could do, essentially, iterated epigenetic CRISPR editing—though it seems at least fairly likely that the epigenomic disruption from cell culturing would be worse than the progress you make with editing. This would probably work for only at most a small number of edits, but could be useful in hybrid EC-making.
- Even if you can get monoallelic edits, in theory you could still have problems if you aren't targeting a specific chromosome. Suppose you make the right epigenetic edit on exactly one chromosome. Plausibly you'd thus get your desired trans-regulatory effects (i.e. long-range regulation, e.g. one gene makes a transcription factor that diffuses around and binds and regulates some other gene on some other chromosome). But you might not get your desired cis-regulatory effects (i.e. effects on nearby DNA, e.g. you methylate a promoter and thereby suppress expression of the gene next to that promoter). Plausibly there wouldn't be any missing effects, e.g. because the DNA would be mostly identically regulated except at a few places, none of which have cis-regulatory interactions with each other.
- Diploid-to-haploid epigenetic editing: You establish biallelic maternal imprinting or biallelic paternal imprinting. Then you somehow derive a haploid without disrupting the state.
- E.g. possibly you could induce meiosis, though this would probably disrupt the state.
- E.g. possibly you could do chromosome selection of some sort.
- I'm not sure if there's ever a benefit to this, over just haploid editing. One possible benefit is that the diploids might be easier to culture than the haploids, making iterated epigenetic CRISPR editing easier.
Challenges:
- CRISPR-based epigenetic editors have many of the same problems as CRISPR-based DNA editors. They generally will have off-target effects—e.g. they methylate regions that weren't targeted[87]. Depending on the variant, they might have low efficiency (they don't make the target edit). They also might be difficult to deliver to the target cells if the cells are embedded in some tissue (e.g. germline-cell-like cells in gonadal tissue).
- Epigenetic editors whose function relies on cellular machinery wouldn't work if the cell being edited doesn't have that machinery. E.g., sperm-like cells would tend to be missing a lot of normal machinery because they are mostly inactive cells.
- Full EC-making would be difficult using epigenetic CRISPR editing alone.
- In other words, it would probably be hard to take an hiPSC (haploid or diploid), apply some epigenetic editors, and get a cell that can be used as a zygote nucleus or as a uniparental contribution.
- Full EC-making would require not only establishing sex-linked imprints, but also general epigenomic reprogramming. E.g., PGCs become broadly demethylated compared to normal ESCs, and gametes become very silenced. This would have to be accomplished some way other than a huge number of single locus epigenetic edits.
- Safe full artificial EC-making, in contrast to natural EC-making, would require knowing in great detail the necessary epigenetic state.
- With EC-making that is natural or natural-like (e.g. induced in vitro), the imprints are established via something like the natural cell pathways. Those pathways would tend to do all or almost all of the reprogramming needed, somewhat by default, even if you don't know about some of the needed imprints. In contrast, with epigenetic editing, the most upstream epigenetic imprints are each individually likely to be absent unless you specifically make those edits.
- If the goal is to make a haploid genome, the cell would have to be meiosed somehow. This would separately require a method to induce meiosis. If you did the EC-making before meiosis, meiosis might mess up the epigenetic state.
So, epigenetic editing seems more likely to be used in hybrid methods than as a full EC-making method. It could be useful to e.g. fix a few imprints that were lost while culturing naive ESCs or that failed to be established by an imperfect IVG method.
As an illustration of the principle, though not directly an example of epigenetic CRISPR editing: Li et al. (2025) made bi-paternal mice by making genetic edits, targeted to genes affected by sex-linked imprinting, aiming to correct the activity of those genes. This was partly successful, in that they greatly decreased the rates of various abnormalities, and produced live births; but only a few made it to adulthood[16:1].
A more direct illustration, mentioned above: Liu et al. (2018) cloned cynomolgus macaques with SCNT[9:2]. They altered the histone modifications in the DNA before transferring it to enucleated oocytes. This greatly improved their SCNT success rates (though they still had epigenetic near-misses, as mentioned above).
Donor embryo
This is a method for lowering the required EC bar. You don't ask for a cell that can form the entire conceptus, including the fetus and placenta and amniotic sac, as a normal zygote can. Instead, you take a donor embryo and somehow disable it from growing a fetus. Then you inject your GVed cell or embryo into the donor conceptus. The injected cells take the fetal role, while the donor grows the placenta and amniotic sac. See for example the VelociMouse method[88].
The main benefit of this method is not exactly about imprinting, but rather about the pluripotency of your GV cells. Naive ESCs can form the whole conceptus, but primed ESCs can only form the fetus and not the trophectoderm. Naivety is harder to maintain in culture than a more differentiated state. By using a donor embryo to provide the trophectoderm-descended tissues, you don't need to maintain naive pluripotency.
Another part of the hope with a donor embryo is that most sex-linked imprinting is about the placenta, rather than the fetus. So, even if the progenitor still requires some EC-making, the amount of EC-making you still have to do might be greatly decreased. For example, it might require fewer epigenetic edits.
I don't know much about this method. I suspect it's not straightforwardly viable because sex-linked imprinting is not just about the trophectoderm. It also might produce mosaicism. The concern about mosaicism might be largely avoided by using a twinned embryo as the donor.
Hybrid natural and artificial EC-making
It might be that we find a method to partially but not fully correct the imprints of some DNA artificially in vitro. In that case, we could compose that method with natural epigenomic correction.
Examples:
- Natural EC DNA and epigenetic editing. EC bypass methods that want to just use naturally EC DNA don't necessarily fully preserve imprinting. E.g. culturing and editing ESCs or SSCs would, by strong default, lead to some loss of imprinting. But if they mostly preserve imprinting, the remaining aberrations could maybe be corrected with a small number of epigenetic edits.
- Spermatogonial stem cell induction. If we could artificially make iSSCs, we could apply GV to iPSCs, induce iSSCs, and then transplant them. This is a genuinely hybrid EC-making method because SSCs already have some paternal imprinting; our induction method would have to make those imprints, as well as general epigenome-wide reprogramming.
- In vitro spermatogenesis proper. Conversely, suppose we find a method to take natural SSCs and induce them into spermatogenesis in vitro, i.e. meiosis and further epigenomic reprogramming. In this case, we could extract adult SSCs, apply a GV method, and then use IVS. The GV method would have to preserve the SSC imprinting that is supposed to be transmitted to male gametes.
- Immature oocytes.
- In theory, it might be possible to retrieve oocytes from ovaries that are not yet at the germinal vesicle stage with preantral / antral follicles. We could apply a GV method to these, and then apply in vitro maturation to complete the oocyte growth and imprinting, and undergo meiosis.
- This is probably infeasible, as there's no way to get such immature oocytes in general.
- At first this hypothetical method seems entirely pointless. E.g., it doesn't seem especially easier to apply GV to immature rather than mature oocytes. But one potential benefit is that, if there were a method to safely and ethically obtain immature oocytes (e.g. if there are some in adult ovaries), then they might be more abundant than mature oocytes. All oocyte maturation methods that I'm aware of have low efficiency, in the sense that there are many more immature than mature ones. So if the GV method has some attrition, it might be better to apply GV at a stage where you get more chances. (However, I don't see a benefit to using pre-growth oocytes rather than just using the kind of immature oocytes that are abundant in adult ovaries, which have already received all or almost all of their imprints.)
How GV and EC interact
Reproductive genomic vectoring requires a GV method and a method for handling epigenomic correctness. Is that sufficient? If we have a viable GV method and EC method, do we get reproductive GV? Not necessarily.
A simple-ish and wrong model of combining GV and EC
Here's a diagram displaying, coarsely, ways that GV can interface with EC:
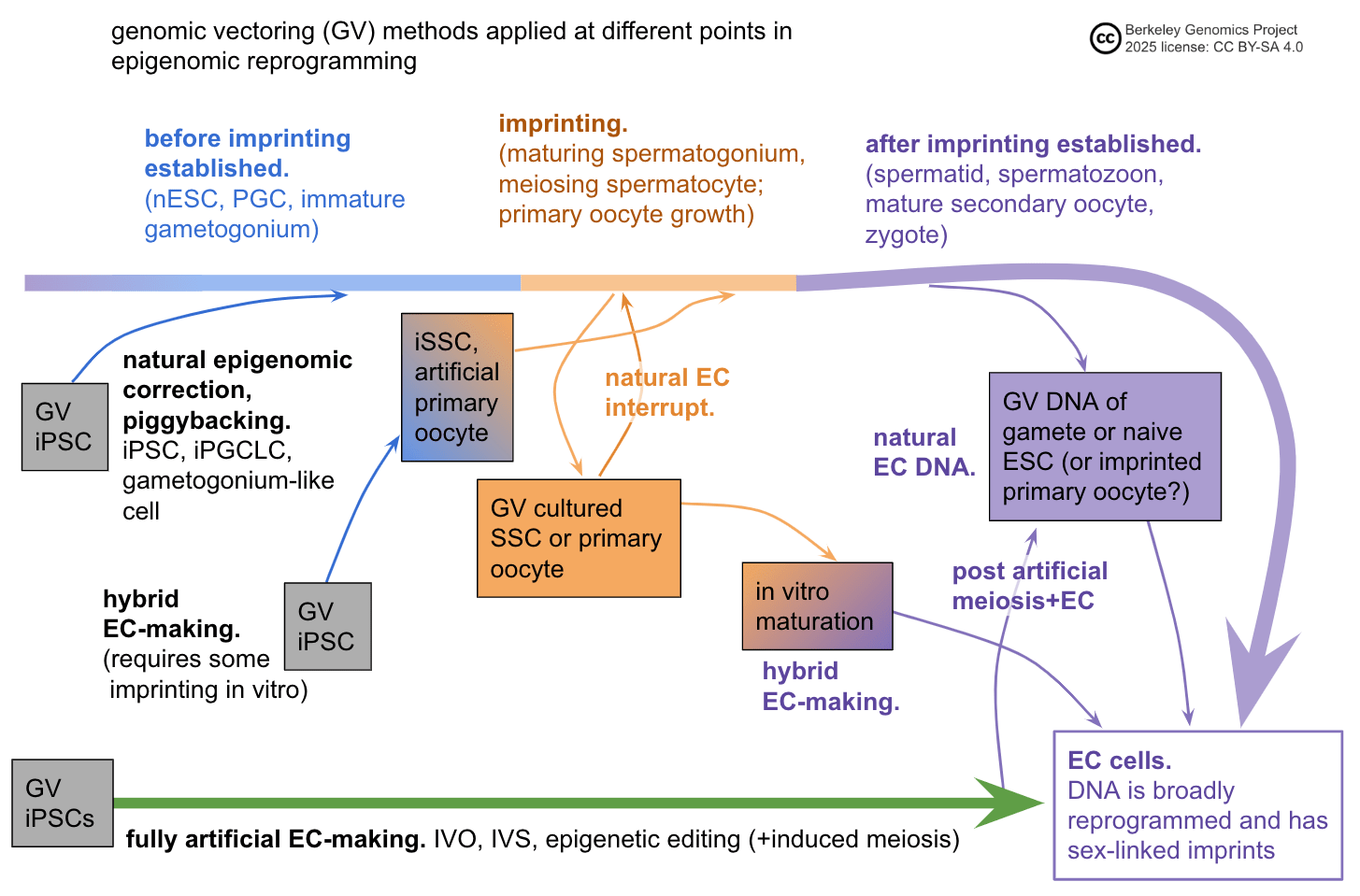
Basically, at a coarse level, you can apply GV at some point in the epigenomic correction process. This could be before imprinting is established; while it's being established; or after it's been established.
GV and EC are not conceptually separable
It would make it easier to think about reproductive GV if we could think about GV and EC separately. Then we could solve GV with some method, and separately solve EC with some other method, and then combine them to get a cell that has been genomically vectored as we choose and that is epigenomically competent for development.
To some extent, this can be done. For example, suppose we had full IVO: we can take any diploid stem cell and make a competent egg. Now, it seems like basically any GV method whatsoever can be precomposed with our IVO method. We take a somatic cell from the parent, induce it to an iPSC, apply our GV method to it, and then use IVO on the GVed iPSC to get a GVed egg.
But there is a caveat missing. It's not technically true that any GV method could be used in exactly this way. Specifically, consider recombinant chromosome selection. The power of recombinant chromosome selection is that recombined chromosomes can be higher scoring than either of the two parent chromosomes. So it only makes sense to do recombinant chromosome selection after meiosis. But taken literally the above protocol would say to "do recombinant chromosome selection on iPSCs, then do IVO", which doesn't make sense, as iPSCs are diploid and are genomically the same as the adult whose cells they're derived from.
This isn't some great difficulty with recombinant chromosome selection. You could probably, for example, do chromosome selection to some artificial eggs to make a diploid; make it an iPSC; and do IVO again to get a high-scoring, EC oocyte.
My point is rather that, in general, we can't just separate out GV from EC.
More examples to illustrate the point:
- Chromosome selection might be doable on sperm DNA. However, iterated editing almost certainly can't be done on sperm DNA: sperm don't have the requisite cell machinery, sperm DNA is highly inaccessible, and sperm can't be cultured for iteration. Thus, the general EC bypass method "just use natural sperm DNA" is only compatible with some GV methods. Likewise for natural oocytes, as oocytes have already started meiosis and therefore can't normally be cultured.
- On the other hand, if naive ESCs can truly be cultured such that they would still form viable embryos, then both editing and chromosome selection ought to work with naive ESCs. So some EC bypass methods could work with both editing and selection methods.
- Suppose you can culture adult spermatogonial stem cells in a way that maintains their imprinting. Then, you can do iterated editing to these SSCs, transplant them back into the adult's testicles, and retrieve edited spermatozoa. Can you also do chromosome selection? On the face of it, yes; chromosome selection is supposed to be minimally disruptive of epigenetics, in theory. However, chromosome selection requires access to individual cells, separated from their context. If the epigenomic state of SSCs is fragile, this separation would be disruptive. So chromosome selection may not be compatible with this EC method. Then again, editing also wants to separate the SSCs for a little while in order to verify edits and non-damage; but then again again, maybe that step can be deferred to a single screening round of embryos after retrieval and fertilization.
- Different implementations of broad methods have different epigenomic consequences. For example, chromosome selection that uses some kind of mechanical manipulation might be fast enough to not damage epigenomic state. On the other hand, poor man's chromosome selection involves several rounds of mitosis and exotic tetraploid states, so it seems unlikely to preserve epigenomic state by default. Both would work on iPSCs followed by IVO; but only mechanical chromosome selection seems plausibly compatible with using natural SSCs.
Features of GV and EC methods that affect compatibility
A given GV or EC method is, vaguely speaking, some strategy for applying some manipulations to some cells or DNA. Here are some aspects of GV or EC manipulations that affect which other methods they can and can't be combined with:
- Ploidy.
- Some methods have to work with haploid cells, and some have to work with diploid cells.
- IVG methods probably can't apply straightforwardly to haploid cells.
- E.g. if poor man's chromosome selection works via spontaneous tetraploid reduction, then it has to work with diploids.
- E.g. epigenetic CRISPR editing to make a gamete has to work with haploids (unless imprints can be preserved across meiosis).
- Methods can preserve (e.g. mitosis) or change (e.g. meiosis, cell fusion) ploidy. Most IVG protocols probably can't perform imprinting without also turning diploids into haploids.
- Meiosis is not necessarily the only way to make haploid cells. For example, some forms of chromosome selection might allow one to make a haploid "manually". E.g. if you could specifically destroy several target chromosomes without totally destroying a cell, you could destroy a haploid genome, leaving the other haploid genome.
- GV methods applied before diploid-haploid reduction will generally be half as powerful (if the haploid is used).
- Position relative to meiosis.
- By default, EC-making methods have to either come after meiosis, or come along with meiosis (most IVG), or have a way of preserving imprints across artificial meiosis.
- Recombinant chromosome selection has to come after at least one step of meiosis.
- Culturability.
- Some methods can only work with culturable cells.
- Generally iterated methods, such as iterated editing, iterated meiotic selection, and poor man's chromosome selection, only work with cells that can be grown.
- Mechanical chromosome selection could work with non-culturable cells.
- Some methods produce non-culturable cells.
- Generally, meiosis produces haploid cells, which might not be easily culturable.
- EC-making methods might produce cells that can't be cultured by default, but rather can only be cultured if there's an additional method to maintain imprints.
- Fully EC cells are heavily silenced, and therefore can't be cultured.
- Most IVG methods produce cells with naturally very silenced genomes, which therefore can't be cultured without further intervention.
- Some selection methods might produce intermediate diploid cells with a lot of homozygosity, which might be unable to mitose or might be unsafe to mitose (because you're applying some weird selection pressure).
- Some methods don't automatically preserve epigenomic correctness.
- Cell access.
- Some methods require total access to isolated cells in vitro, some require less total access.
- Editing doesn't require total access. E.g. one could edit SSCs that are growing inside testicular tissue in vitro or even in vivo. (However, without at least one step of total access for screening, some DNA damage accumulates; so you have to screen embryos, which is costly.)
- Most selection methods require total access.
- Some methods provide access, some don't.
- Cell stress.
- Cells get stressed by damage. Then they aren't as good at proliferating or other activities.
- Most GV methods would stress the GVed cell.
- EC methods can be more or less compatible with stressed cells.
- For example, I would guess that sperm being stressed is not so bad. Sperm are mostly inert anyway, and are dissolved upon entering the oocyte, so they don't have much responsibility to add to their stress lol. Therefore, e.g. sperm selection (using natural EC DNA as an EC bypass method) should not be a problem in terms of stress.
- On the other hand, applying a GV method to SSCs and then retransplanting them into testes would pit the GVed SSCs against the natural SSCs already in the testes. If the GVed SSCs are stressed, then by default they would likely be outcompeted by the other SSCs.
- Some methods such as genetic or epigenetic CRISPR editing require some machinery to be present in the cell, e.g. methylation enzymes.
- Some methods require certain chromatin states.
- Chromosome selection requires that the chromosomes be condensed enough to manipulate, but not entangled with or bound to each other (as in meiosis).
- CRISPR editing and DNA labeling require that the chromatin be accessible.
How feasible are different epigenomic correctness methods?
Most of the following judgements are speculations. They're based on reading what biologists write, but many of them are just guesses; they're intended as a snapshot of my current understanding, and to give experts something to disagree with.
These are arranged in rough order of easiest to hardest, in terms of getting them to work as methods of handling EC (but not necessarily as strong GV methods).
- Just use normal reproductive DNA.
- E.g. simple embryo selection, sperm or oocyte chromosome selection.
- Just use normal reproductive DNA, with a short window of operating on the DNA.
- E.g. a small amount of editing on ESCs; ESC chromosome selection.
- Use natural epigenomic correction.
- E.g. in vivo spermatogonial editing; spermatogonial stem cell induction and testicle transplantation; oogonial stem cell induction followed by oocytogenesis and maturation in ovarian tissue in vitro.
- Imprint maintenance and normal reproductive DNA, plus a small amount of epigenetic editing.
- E.g. editing ESCs for a longer period of time.
- IVO.
- IVS.
- Donor embryo. (I'm guessing donor embryo methods are easier to make sort-of work, but also more likely to be a near-miss.)
- Massive epigenetic editing to directly create a gamete or embryo.
- Creating an embryo directly from a stem cell in some other way.
How epigenomically disruptive are different GV methods?
Note: The following judgements are speculations, like the ones above about feasibility of EC methods.
No epigenomic disruption
These methods might not disrupt epigenomic state at all:
- Simple cell population selection
- Mechanical chromosome selection
- Single-round multiplex CRISPR editing
Thus, these methods can use EC bypass methods, e.g. simple embryo selection (using already EC cells), editing zygotes, or sperm chromosome selection. These methods are most likely to be feasible now or in the near future.
Little epigenomic disruption
These methods might not disrupt epigenomic state too much:
- Iterated multiplex CRISPR editing
- This method requires several weeks or months to operate, and requires cells to undergo mitosis. During that time, by default, special epigenomic states such as being a naive ESC or being a spermatogonium will degrade, e.g. with loss of sex-linked imprinting or loss of stem cell potency.
- Chromosome selection via chromosome digestion and electrofusion
- This theoretical method might totally destroy cells or massively disrupt their state by setting off lots of DNA damage response mechanisms. But if that can be circumvented somehow, the intact chromosomes might be mostly unaffected.
So these methods can take advantage of weak EC-making methods, e.g. small numbers of epigenetic CRISPR edits or transplanting vectored gametogonia back into in vivo gonads.
Significant epigenomic disruption
These methods probably disrupt epigenomic state a lot:
- Very many rounds of iterated multiplex CRISPR editing
- Iterated recombinant selection
- Chromosome selection via whole cell fusion and tetraploidy reduction
Many rounds of CRISPR editing involve even more cell culturing, which by default disrupts epigenomic state. For example, if culturing takes a couple weeks, and you can get 20 edits per round, and you want several hundred edits, that takes over half a year. That said, it may be feasible to find methods, in the vein of super-SOX, to strongly maintain the needed epigenomic state in culture, in which case strong iterated editing would work.
Iterated selection, e.g. IMS, involves inducing meiosis. That process presumably has significant epigenomic effects, including some loss of imprinting. More importantly, because IMS shuffles chromosomes and segments of chromosomes between cells, it will mix and match chromosome segments with paternal and maternal epigenetic imprinting. This means that IMS absolutely needs a strong EC-making method.
Tetraploidy may not cause further significant epigenomic disruption even with methods to maintain imprinting, so it may require strong EC-making methods.
Haploid vs. diploid
Some ways of handling epigenomic correctness allow for a diploid GV genome to contribute to an embryo; others only allow for a haploid contribution.
In general, a haploid GV genome contribution has half the GV power as a diploid contribution:
- For editing, most edits will be fairly common variants (minor allele frequency >1%, say). For such edits, in most cases it's fine to edit both chromosomes. Homozygosity shouldn't be a problem for common variants. So usually a diploid contribution can take twice as many edits as a haploid one, giving twice the effect, for roughly the same cost (most of the cost is one-time-per-edit-type, e.g. figuring out a good guide RNA to use).
- For selection, the effects simply add between the two halves of the genome. (That is, selecting half a genome is half the effect of separately selecting both halves. However, selecting half a genome is not half the effect of selecting a whole genome, but rather is times the effect, i.e. roughly 70%.)
Which ways of handling the EC problem enable haploid vs. diploid GV?
- In vitro gametogenesis is a haploid EC-making method. Just because you can do in vitro oogenesis, doesn't necessarily mean you can do in vitro spermatogenesis, and vice versa. So you only get haploid GV power. (If you have both IVO and IVS, then you probably always get diploid GV power.)
- Natural EC-making likewise gives haploid GV power.
- Epigenetic CRISPR editing ought to be diploid, though I don't know. If you have epigenetic editing methods working well enough, you ought to be able to correct for both paternal and maternal imprinting; the two types of imprinting have roughly similar complexity to each other. On the other hand, it might be harder to know the target state for sperm or for eggs, or harder to validate one or the other with trials.
- Using natural reproductive DNA ought to give diploid EC cells. If some GV method can avoid disrupting the epigenomic state, it likely would apply to both maternal and paternal DNA. But, this is far from guaranteed. For example, sperm DNA might be more difficult to work with than oocyte or embryonic DNA, because it is highly condensed.
Summary of genomic vectoring methods
A genomic vectoring method aims to create a cell that has a haploid or diploid complement of chromosomes that's been somehow steered (selected, modified). The following sections go into detail on several GV methods—the basic method, the obstacles to implementing it, and estimates of its genomic vectoring power. They are ordered roughly in terms of how hard it is to analyze the method's vectoring power. Simple embryo selection is the simplest to analyze coarsely; iterated recombinant selection is the most complicated selection GV method; and editing is not something I've worked on, so I'm deferring to others.
An annotated table of contents for the following Method sections:
- Simple embryo selection.
- Make embryos, select the high-scoring one.
- This is a weak GV method.
- This is already being done.
- Gamete selection.
- Select the high-scoring sperm and/or the high-scoring egg, and combine them.
- This would be a fairly weak GV method.
- This might become doable given non-destructive DNA sensing.
- Chromosome selection.
- Select the high-scoring chromosomes from several cells and put them together in one cell.
- Surprisingly strong. Chromosome selection on embryonic DNA from one couple would give over 6 SDs of raw selection power.
- Requires some form of DNA sensing. The hardest part is getting the target chromosomes together in one cell, intact.
- Has a good chance at bypassing the EC problem by using already-EC DNA, so is fairly uncorrelated with the success of almost all other reproductive GV methods.
- Iterated recombinant selection.
- Induce diploid cells to divide meiotically into haploids, then combine the haploids to make new diploids, selecting high-scorers along the way.
- This would be very strong GV.
- This requires directly inducing meiosis, and requires an epigenomic correction method.
- Iterated multiplex CRISPR editing.
- Apply multiple rounds of multiple CRISPR edits to lines of stem cells.
- Given an epigenomic correction method, editing is very strong; without one, editing might be anywhere from weak to strong (by editing ESCs with epigenomic maintenance).
The following table summarizes the genomic vectoring power of the main GV methods. It's ordered roughly in terms of how feasible I think the method is—specifically, how soon I think it could be done, if someone seriously tried, soonest first. These estimates are uncertain and involve lots of modeling assumptions, but they're probably in the right ballpark.
[(Link to spreadsheet, which you could inspect, copy, and modify: https://docs.google.com/spreadsheets/d/1AaBs7d772q_UgReIMCYM3m1-Jc3QHvcBhfg47gsK0ug/edit?usp=sharing. I have found (and epigenomically corrected) several errors in drafts of this spreadsheet, so please be on the lookout.)]
The order statistics given at the top, which are used in many of the power estimates for selection methods, can be checked with this snippet:
import numpy as np
def expected_max(test_runs=10_000_000, k=2):
X = np.random.randn(test_runs, k)
return np.mean(np.max(X, axis=1))
All the IQ point numbers are additional over the mean population score , and assume that all input genomes are drawn from the population at random, with a standard normal distribution centered at . The mean can be whatever you want, though at the very high end there is a ceiling on IQ (it's not known where or with what distribution).
This table is only a summary. More analysis is given in the following sections. There are many combinations of GV methods and EC-making methods that aren't specifically covered, though in general combining GV methods would significantly increase genomic vectoring power.
Except for simple embryo selection, all of these methods are speculative, at least in humans. No one has done them yet, and furthermore they all require scientific and technological understanding that hasn't been achieved yet. They will likely have many unforeseen obstacles (though will also in some cases be easier than expected).
All the discussion below focuses on making a single child. See "Selection makes similar children" for a potential issue with this assumption.
Method: Simple embryo selection
Method:
- Do in vitro fertilization (IVF) to get some embryos.
- Sequence the embryos by taking a few cells from each.
- Pick the highest-scoring embryo to try to implant.
Simple embryo selection is already being done; see for example Orchid, Lifeview, Heliospect, and GenEmbryonics.
The power of simple embryo selection
The effect of embryo selection is severely limited by two factors:
- IVF produces only a small number of achievable births, i.e. eggs produced during superovulation which are successfully fertilized, develop normally enough in vitro, and then successfully implant and gestate. An older women might obtain only a few or even no embryos; with multiple superovulation rounds and good reproductive health, a young woman might optimistically hope to have one or two hundred achievable births to select from.
- The variance in the genomes of embryos is half the population variance (the variance of a random person's genome).
Together, these effects severely limit the practical effects. Assuming a PGS that correlates at 0.4 with IQ, we get:
This graph uses a simple model, where the embryos for one couple are drawn from a gaussian with variance roughly .5 (more exactly, .496). To give some numbers:
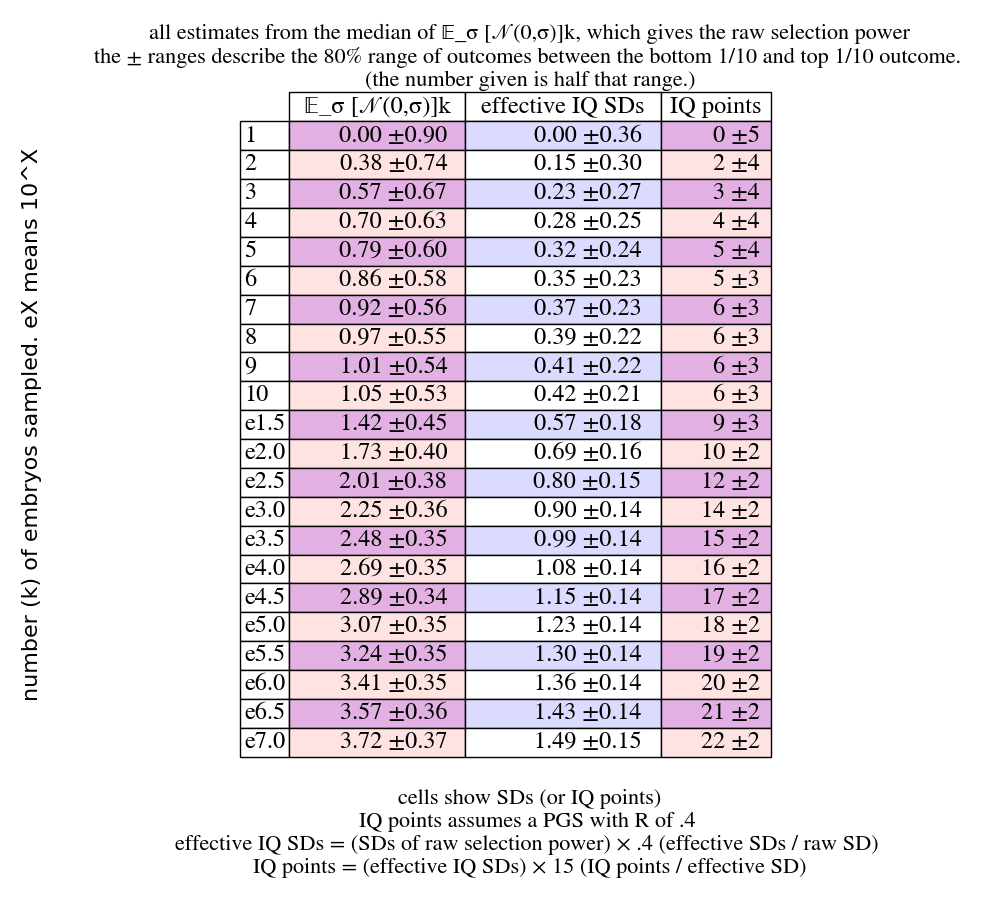
This table shows the gains on IQ if you select the top-scoring embryo out of embryos, where is shown in the leftmost column. (In what follows, in the context of IVF, really means achievable births.) The leftmost column is ; the next column shows raw SDs of selection power. The next column shows effective-SDs or trait-SDs for IQ. Raw selection power is just "how uncommon is this embryo"; the effective selection power or trait selection power is "how uncommon is this embryo, on the scale of this trait". If the PGS for IQ correlates with IQ at 0.4, then a 1 SD exceptional embryo on our PGS will be around 0.4 SDs exceptional on actual IQ.
(Different couples have different expected gains, though not due to their average IQ. The distribution used in the table above integrates over the uncertainty over the couple's embryo-variance , and the values given are the median with an error range spanning 10th percentile to 90th percentile. Also, the median values can be fairly easily approximately calculated as follows: take the max-of- distribution for a standard gaussian, given in "Samples to standard deviations"; and then divide the given number by , since the variance of one couple's embryos is the variance of the general population. E.g. a rarity of in corresponds to 4.8 SDs; ; which is what the above table gives for e6.0. But the table also gives a sense for the range of likely outcomes, which is harder to estimate. See the appendix "Detailed estimation of embryo selection".)
We quickly hit a big wall when sampling more and more. Going from 1 to 100 embryos gives +10 IQ points; going from 100 to 1,000,000 also gives +10 IQ points. We don't get very far with realistic achievable births with current methods:
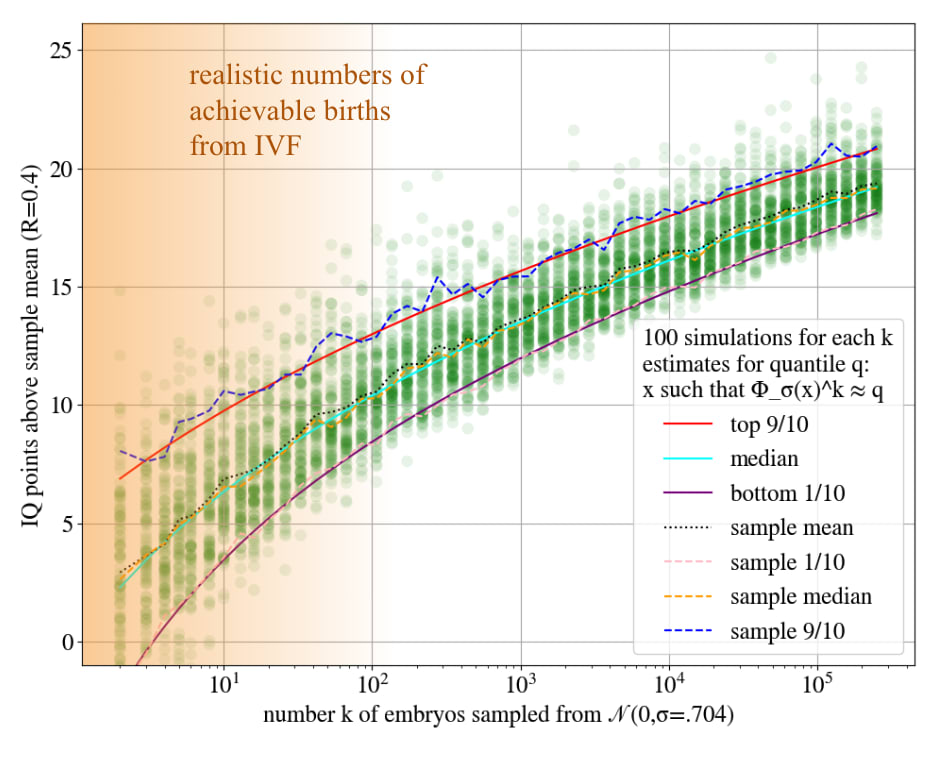
Super-duper-ovulation
Several research groups are attempting to work out a method for in vitro oogenesis—making eggs artificially by taking stem cells and stimulating in them the cellular processes that turn them into eggs. If one of those groups succeeds, you could generate large numbers of eggs, fertilize them, and then apply simple embryo selection.
It might also be possible to harvest many eggs via ovarian tissue extraction, stimulation of dormant follicles[63:1], and in vitro maturation. Ovarian tissue cryopreservation is a treatment practiced to restore fertility after chemotherapy, so we know that ovarian tissue can produce eggs after having been extracted[89]. Human oocytes can be fully matured from the adult stage (arrested in prophase I; antral follicle) to an oocyte, in vitro[64:1]. However, the current practical efficiency of these methods is not great[90].
In principle this method might be able to produce many natural eggs in order to make many more embryos than is possible via superovulation. However, normally as follicles grow and oocytes mature in preparation to be ovulated, they suppress each other until one is dominant and is ovulated. This ovarian follicle dominance might stop you from getting, say, hundreds or thousands of eggs from extracted ovarian tissue. In the most successful case, this would be limited to under half a million eggs, as adult women have at most that many oocytes with primordial follicles.
One could imagine getting tens of thousands of eggs, or more. This would be a 50% to 100% improvement over simple embryo selection, which is still not very strong.
You would want to use some extremely cheap screening method, to avoid paying millions of dollars in sequencing costs. Batched small SNP arrays might be cheap enough, or if not, some more complicated method might work; the appendix "Cheap DNA segment sensing" discusses this question. If you used FISH, you'd just pick the embryo whose biopsy was the brightest. (Or rather, pick the brightest 10 or 50, and then whole genome sequence them and pick the highest scoring one.)
Method: Gamete selection
Single gamete selection
Method:
- Collect a bunch of gametes (either a bunch of sperm or a bunch of eggs).
- Somehow find a small subset of those gametes that are higher scoring.
- Combine those gametes with complementary gametes, producing embryos.
- Sequence the embryos and pick the highest-scoring one to try to implant.
Gamete selection is mostly hypothetical
The basic problem is that all high-information DNA analysis (sequencing, SNP genotyping) is destructive—to sequence a cell, you destroy it, amplify the DNA, and then apply a lossy detector of some sort. Further, by default there's no way to culture a gamete. Both sperm and eggs are in a kind of stasis; their DNA is largely epigenomically silenced, so they don't produce the proteins that would be needed to mitose.
In theory one could do "entanglement sequencing", wherein you capture the four meiotic "grandchildren" of a single gametogonium (progenitor stem cell that produces gametes). This is impractical as far as I know, because both oogenesis and spermatogenesis are complex and all but require a bunch of surrounding tissue. Spermatogenesis in particular involves a complicated process where many partially-differentiated progenitor cells stay tightly connected as they pass through the seminiferous tubules. Oocytes might be more feasible to indirectly sequence, e.g. by looking at the polar body emitted after false fertilization.
In any case, there's little point in doing oocyte selection unless you either also have sperm selection, or you're more constrained on processing embryos than on producing oocytes. Otherwise you're just going to fertilize all your oocytes with a random sperm anyway and then sequence it, so it doesn't really matter if you know the oocyte's haploid genome beforehand.
But sperm selection might be doable
What you'd need is some method for non-destructively determining enough about a sperm's haploid genome, that you can sort many thousands or millions of them. I don't know of a method that clearly ought to work.
Something like FISH might work. You would label several variants that appear in dad's paternal chromosome but not in his maternal chromosome, or vice versa, depending on which segment is higher scoring. See the appendix "Cheap DNA segment sensing". Then you'd pick the brightest or most multicolored sperm, e.g. using FACS.
The power of single gamete selection
How many SDs of selection pressure do you get from single gamete selection?
Variance is additive, i.e. . The variance of an embryo is the sum of the variances of the gametes from each person. So the variance among the gametes sampled from one person is half the variance of the embryos of a couple. In terms of the effects of selection, everything scales in terms of SDs . The SD of a gaussian with variance is , i.e. the square root of the variance. So the gametes drawn from one person, which have a variance that of embryos drawn from a couple containing that person, will have a standard deviation of .
We can use this to convert from the numbers for simple embryo selection to numbers for gamete selection. If we only did gamete selection, taking the top-scoring of gametes, and didn't select the resulting embryos after fertilization, the selection powers would be times whatever the power would have been for selecting embryos.
Double gamete selection
Suppose that sperm selection works, as described above. We'd then combine sperm selection with embryo selection. Once you have one or several top-scoring sperm, you'd fertilize some eggs to obtain embryos; sequence them; and pick the highest-scoring ones.
We can think of this as essentially being double gamete selection, which in general is the following method:
- Get a bunch of sperm, pick the highest-scoring one.
- Get a bunch of eggs, pick the highest-scoring one.
- Fertilize the egg with the sperm.
Doing sperm selection followed by embryo selection is not quite the same power as double gamete selection. Instead of pairing the very best sperm with the very best egg, you pair the top, say, 20 best sperm with 20 random eggs, and then pick the best; you probably didn't pair the best sperm with the best egg.
On the other hand, if you select the top 20 from millions of sperm, they'll mostly be around what you expected to get from the top 1. Then when you select your embryo, you add on the power of selecting the best egg. So the loss compared to actual double gamete selection is not necessarily very noticeable. (Technically, you do better than best egg plus top 20 sperm, because maybe the second best egg was paired with the top sperm, and together they are higher-scoring than the best egg paired with another sperm.)
Thus we can roughly view sperm selection followed by embryo selection as being double gamete selection. Further, suppose we have in vitro oogenesis. Then it would become worth it to do oocyte selection as well as sperm selection, getting the full power of double gamete selection.
The power of double gamete selection
If we abstract away those details and consider the mathematical situation, how many SDs of (raw) selection power does double gamete selection give?
Using the discussion above, we can give a naive answer. (See "The power of selection" for a more convoluted discussion[91].) If we combine the single best of sperm with the single best of eggs, we get something that is times more SDs than the single best of embryos from the same couple. Call the latter value , meaning "expected maximum of samples from the (mbryo) distribution", and likewise for (ametes). So we have
and in general,
So for example, suppose you can use in vitro oogenesis to make 1,000 eggs, and you can select from 10,000 sperm. Then you look up the values in the table above, reproduced here:
We have and . So we get
SDs of raw selection power. Multiplying by the PGS strength of .4, we get 1.4 SDs of IQ, or 21 IQ points. Pretty cool, though not quite strong GV.
The limits of extreme gamete selection
This simple model will break down. The distribution of gamete scores is not really a gaussian. There's some distribution induced over single chromosomes by the crossover process; then 23 of these variables are added together to get the haploid genome of a gamete. Adding together a bunch of variables kinda makes a gaussian, but not exactly.
The upshot is that outside of a few SDs, gamete selection basically hits a wall, defined by whatever the best crossovers for every chromosome are. I think this wall is somewhere around 9 SDs of raw power? I'm not sure. That guess is based on the best crossover being about standard deviations (see the appendix "Best crossover"); applying that to each of 46 chromosomes, each with 1/46 of the variance, gives . But there are other relevant variables not accounted for here. For example, there may be quite a large variation in how many crossovers there are in a given gamete. E.g. a 99th percentile crossover-count sperm might have as many as 37 crossovers, meaning many chromosomes will have a few crossovers[92]. That raises the ceiling a lot. In particular I'm not sure when the gaussianity assumption materially breaks down. However, at a wild guess I'd think you could get to +6 SDs of raw selection power, or +2.5 SDs of IQ, i.e. around 50 IQ points. Not bad, if you can select from huge amounts of gametes.
Method: Chromosome selection
Method:
- Take one or more cells.
- Get enough information about their genomes to identify target chromosomes.
- Somehow arrange that target chromosomes from different cells make into one cell.
- Use that cell to make a baby.
An illustration:
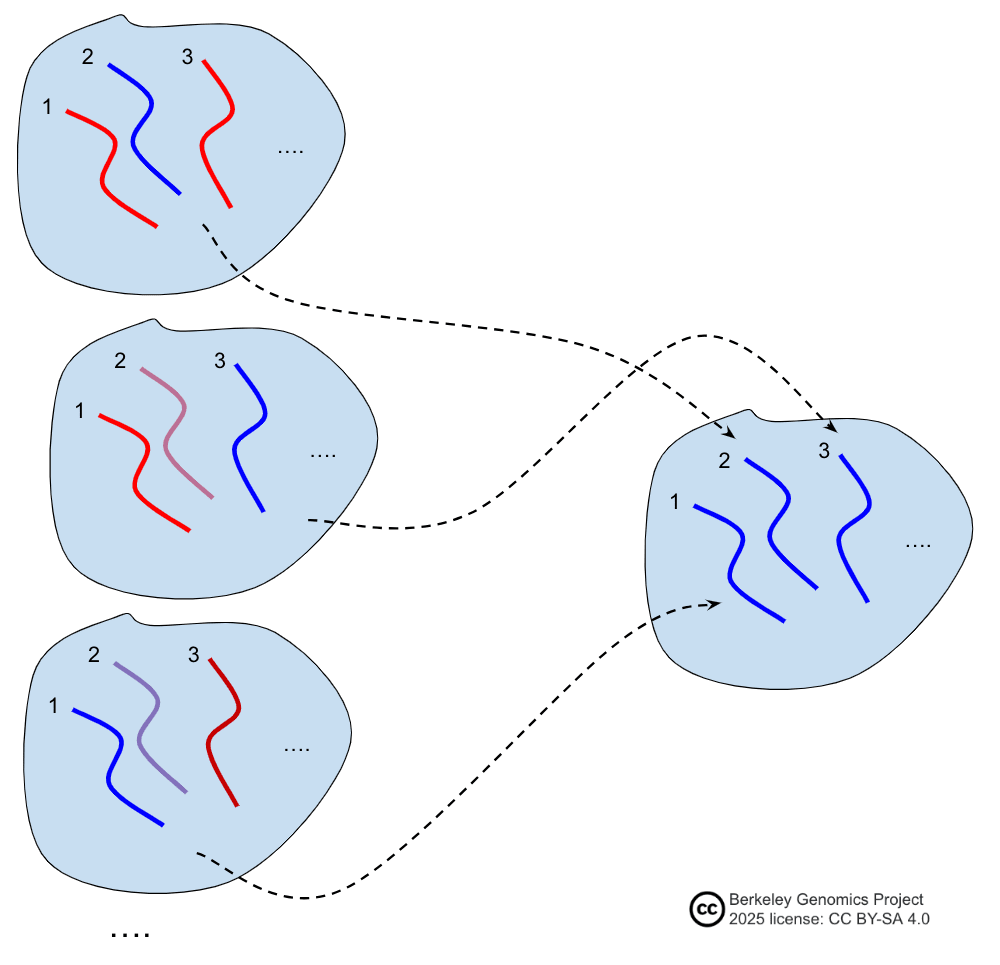
The earliest discussion of chromosome selection I'm aware of is this comment, referenced by Branwen[1:1].
Two key points about chromosome selection:
-
Both average chromosome selection with many donors, as well as diploid recombinant chromosome selection, are truly strong GV methods. See below.
-
In theory, some chromosome selection methods don't disrupt epigenomic states.
- Some hypothetical chromosome selection methods just move DNA around physically for a short period of time, rather than e.g. culturing cells for several weeks or months. In theory, if applied to natural reproductive DNA (oocytes, sperm, embryonic stem cells), this should preserve the DNA's epigenomic state.
- Thus, in theory, chromosome selection can bypass the need for epigenomic correction, which is a major obstacle for most reproductive GV methods. This doesn't mean that chromosome selection is easy; rather, it means that the success of developing chromosome selection is fairly decorrelated with the success of developing other methods, because chromosome selection has a different set of obstacles.
- E.g.: you could apply chromosome selection to sperm DNA.
- E.g.: you could apply chromosome selection to SSCs, and then transplant the frankenSSCs back into a man's testes.
- E.g.: in theory, you could apply chromosome selection to fully grown germinal vesicle primary oocytes. You'd put the chromosomes into a donor germinal vesicle primary oocyte in antral follicle, and then transplant the frankenOocyte into adult ovaries or apply in vitro maturation. (The point of this would be to avoid wasting scarce fully mature oocytes. You'd have to start diakinesis somehow, because the diplotene chromosomes are still connected.)
Implementation of chromosome selection
There are several kinds of method for chromosome selection. See a list of possible methods here: "Chromosome selection". In general, chromosome selection requires some way to identify chromosomes. See cheap DNA segment sensing. Some of the methods in the list linked above don't require direct DNA sensing; e.g. targeted chromosome destruction followed by electrofusion might work.
Two example methods:
Mechanical manipulation
One chromosome selection method that is potentially especially feasible is using FISH to identify chromosomes, combined with mechanical transplantation to a cell. You label the chromosomes, lyse the cells, somehow grab the target chromosome, and then inject it into your target cell.
A major potential obstacle to this kind of method is that it could mechanically break the DNA. There's research about using flow cytometry to study chromosomes, including methods to keep chromosomes intact[93]. So maybe it is feasible.
A great benefit of this kind of method is that it ought not to interfere much with the epigenomic state of the chromosome, assuming that the medium is not disruptive.
An obstacle here is identifying the chromosome. FISH may work, but as discussed in "An obstacle with sperm", sperm DNA may be inaccessible. On the other hand, ESC DNA ought to be accessible for FISH (but could be more vulnerable to mechanical breaking). Therefore this method could be used in an EC bypassing way! This should require minimal culturing of the cell—just enough to get many tries, and then to finally verify the integrity of the final zygote—so loss of imprinting should be small, hopefully.
Another major obstacle is grabbing the target chromosome. One possibility is to use a micromanipulator. However, chromosomes are very small and difficult to see in a normal microscope. It might be doable but difficult with fluorescent labeling. Another possibility is to use FISH and FACS, although FACS might fragment the chromosomes due to shear forces. Another possibility is using magnetic beads attached to the DNA, and then magnetically grabbing them. However, magnetic beads are large, so they might hurt the DNA or might collect dozens of homologous chromosomes.
For sperm DNA, there's a further issue: The DNA is highly compacted and maybe tangled together. That means there's a good chance that it's not feasible to straightforwardly grab a chromosome. It might be feasible to decondense sperm chromatin with chemical treatments[94] and then grab some chromosomes.
Immature oocytes are arrested in diplotene stage, before diakinesis. That means homologous chromosomes are still connected to their homologs by chiasmata (recombination points), and sister (copy) chromatids are still connected to each other at their centers (forming little Xs). Possibly you could stimulate diakinesis and then perform a kind of average recombinant selection on crossed-over sister-chromatid pairs to form a secondary-oocyte-like cell. But this is probably too complicated.
Mature oocytes, arrested in metaphase II, may be more amenable to chromosome extraction. You can artificially activate an MII oocyte as though it were fertilized, and it will proceed with meiosis. After a short while the chromosomes should be nicely separated, while still condensed as chromatin.
These problems shouldn't apply to ESCs.
Whole cell fusion
See "Whole cell fusion". (A.k.a. "poor man's chromosome selection".)
The basic idea is to just fuse two diploid cells together, e.g. via electrofusion, to form a tetraploid cell. Then, somehow, you induce the tetraploid cells to spontaneously correct their ploidy, dividing into diploid cells. One can thus iteratedly shuffle chromosomes around, applying selection pressure to keep the target chromosomes, until you get a diploid cell with most of the target chromosomes.
In other words, iterated whole cell fusion and tetraploid correction is a kind of iterated selection, like iterated meiotic selection but without the meiosis.
As a GV method, this might be doable right now, though it might be pretty inconvenient. Mouse tetraploid cells injected into an embryo were found to sometimes spontaneously correct to diploidy, sometimes giving euploid cells[95]. Maybe one could induce this in vitro.
I assume this method would not maintain a naive ESC epigenomic state, so it would separately require epigenomic correction after producing a GV cell. If tetraploidy reduction were feasible, the whole cell fusion protocol for chromosome selection could be tested soon in non-human species.
The power of simple chromosome selection
In simple chromosome selection, the input cell lines are independent of each other—they're from different people, and don't share abnormally much DNA. Examples:
- You start with several people, and take somatic cells from each.
- You start with several embryos, each one from a different couple.
- You start with one person, and take just one sample.
In any case, the chromosomes can be roughly modeled as just being random samples from a gaussian with variance 1/46 the general population variance, because variance is additive.
Selecting a haploid genome
In the simplest case, suppose that we construct a cell that will play the role of a sperm, by assembling a haploid subset of one parent's own whole genome (that is, without meiotic recombination). We assume that this DNA will fertilize a randomly sampled oocyte. How strong is this protocol?
If we focus on chromosomes at index 1, what happens? We select the best of 2 samples from a gaussian. Very roughly, this gives a result 0.5 SDs above the mean (more precisely, 0.56), in terms of just this chromosome's SDs. We do this 22 times, once for each autosome, and a chromosome-SD is about a normal SD. So we get roughly
SDs. This is fairly weak; if we applied all this raw selection power to IQ, we'd get SDs, or 9 IQ points. Adding in a round of embryo selection gets perhaps another .7 raw SDS, for a total of 13 IQ points.
("Hold on,", you might be saying, "chromosomes have quite different lengths; the longest is 5 times as long as the shortest! Doesn't this greatly decrease the selection power, since variance is pretty concentrated compared to if the chromosomes were all of equal length?" That's a good question, but no, it makes very little difference. See the appendix "Variation in chromosome length".)
What would happen if you took a larger set of partial parents, and selected the best chromosomes taken from any of them? If we construct a haploid this way, we get:
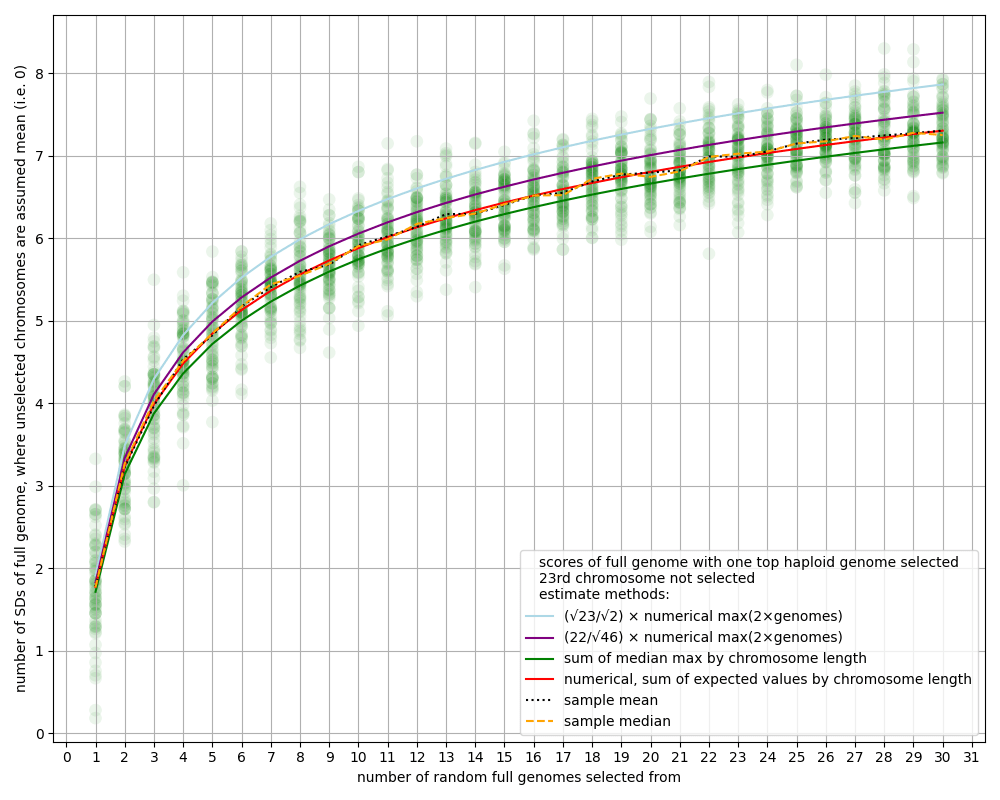
[(This and following graphs are from "The power of selection", which gives more discussion and code for the graphs[91:1].)]
At the bottom left, we see 1.7 SDs for a single genome (best 1 of 2 at each index), roughly as our estimate above said.
Looking further to the right, at 27 on the x-axis, suppose a woman chooses 27 people. Then we sequence those 27 people, and make a haploid from their collective top-scoring chromosome at each index. Then she has a baby gotten from that haploid combined with her egg. Reading the y-axis (the red curve), we see about 7.3 SDs. Combined with a round of embryo selection, that's about 8 SDs of raw selection power. If it's all applied to IQ, that's around 50 IQ points. (If instead she took from 10,000 people, she could get about 80 IQ points for her child, above some combination of the mean of those people's and her own genomic IQ.)
Selecting a diploid genome
Suppose instead that we select the entire genome. There are several possible permutations of this, and I won't go through them all in detail.
E.g., a natural question is, what if you separately create two haploids, one from each parent or one from each set of parents, and combine them? Then you basically add the gains from each. If it's two parents, you get about raw SDs, or about 20 IQ points. If it's many parents, you could do quite well. E.g. with 4 people for each of the two haploids, you can get 9 raw SDs, or about 55 IQ points.
Another kind of question is, what if you create the whole diploid together, by selecting across all the chromosomes? Now you're selecting the top 2 out of some set of chromosomes. Here are the results from that:
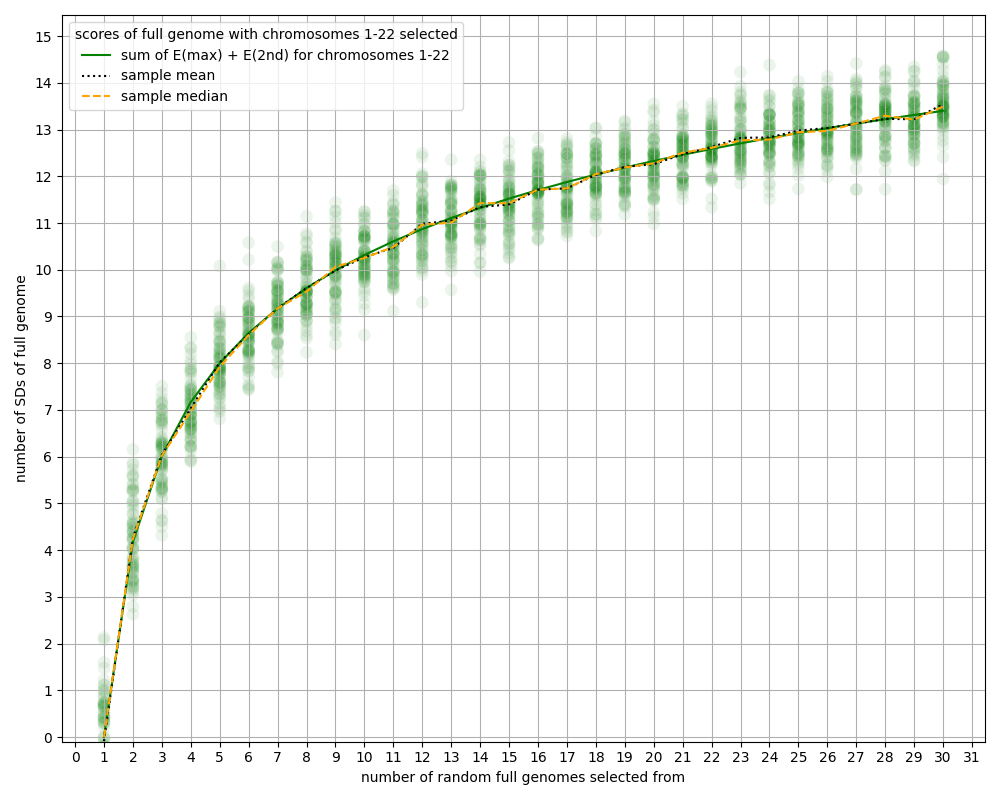
The results are quite strong. (Though, apparently, not much stronger than doing haploids separately—I think this is understandable as "top 1 of " being not that far from "top 1 of ".) Four people can get 7 SDs of raw selection power, or 40 IQ points. (Suppose the same four people want to have several children this way, but without just using the same chromosomes, and keeping the total amount of each person's DNA propagation roughly the same. I don't know how strong this would be, but at a wild guess, we can imagine that the four people could have several children, each with +30 IQ, who are genetically related to all four as genetic grandchildren.)
Another multi-parent arrangement:
- One could replace a small portion of a haploid with chromosomes from other people. For example, we could replace the lowest-scoring quarter of a haploid's chromosomes.
- As a very rough estimate, doing this with, say, around 50 chromosome donors might increase the total raw selection power by an average of SDs per chromosome. If both parents did this with 6 chromosomes, the result would be an additional 6 SDs of raw selection power, or 35 IQ points.
- They'd be genetically related to their children at a closeness halfway between a parent and a grandparent. (One imagines their friends doing similarly, so that no one loses much in total DNA propagation, if that's something they mind. This protocol might, however, potentially have the effect of making their children look noticeably different from themselves—though presumably less different than grandchildren vs. grandparents.)
Selecting an epigenomically correct haploid
Some applications of chromosome selection could bypass the EC problem by selecting from DNA that's already in an epigenomically correct state, rather than applying an EC-making method.
For example, we could start with some embryos from several couples, and then assemble an embryo by selecting a haploid genome of chromosomes with maternal imprinting, and another haploid genome of chromosomes with paternal imprinting. When we do this, we're constrained in which chromosomes to pick for each haploid. Each embryo only contains one chromosome 1 with paternal imprinting, and so on.
In this example, the selection power for starting embryos can be computed by just looking up the power for starting diploids. That's because the set of available chromosomes to be chosen from is halved.
(This example is rather contrived, and is mainly just for purposes of understanding the math. In real life you'd more likely be doing a form of recombinant chromosome selection; see the next subsection.)
Selecting average chromosomes
It might be relatively easy to distinguish chromosomes by index, without being able to distinguish anything else about them. See "Sensing chromosome index".
In that case, given a started set of many donors, you could perform chromosome selection at each index by selecting the person with the highest average score of their two chromosomes at that index. The power of this form of selection would essentially be like chromosome selection, except all the power would be divided by . (The variance of the average of two chromosomes is 1/2 the variance of a single chromosome.) You also have to have twice the number of input genomes to be comparable, because for each index you only get one average-chromosome per genome, whereas you get two chromosomes per genome.
As one example, you could do average-chromosome selection on sperm DNA from many men to create a super sperm. How powerful is this? We can compute this using the analysis in "Selecting a haploid genome". Here's a version of the graph in that section, but with many more input genomes:
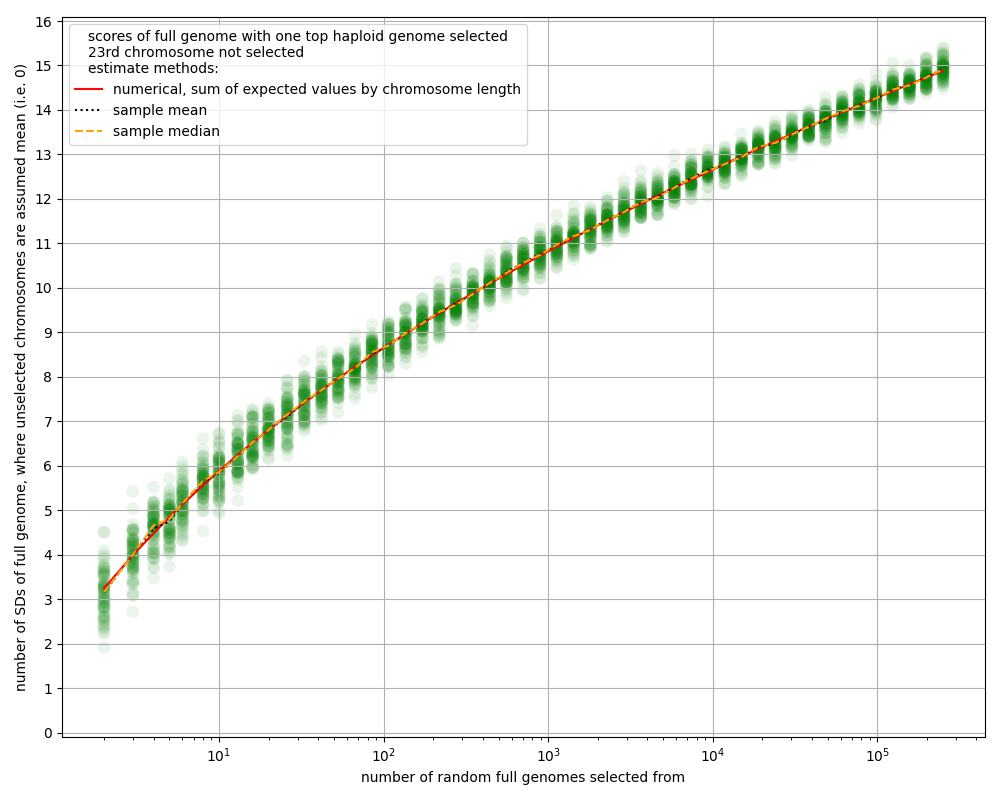
So suppose we have 20,000 men. That's the equivalent of 10,000 whole genomes to select from (because we only get one sample of an average-chromosome per genome). The graph shows about 12.5 SDs of power from chromosome selection. But we have to adjust because the variance of average-chromosomes is less. We get: SDs. That's actually pretty strong!
If instead we take a few hundred men, we get about SDs. Combined with simple embryo selection, that's about 7 raw SDs, or about 40 IQ points. Not bad either.
Can this really be right? We can double check by computing the same thing a bit more directly. With around 200 samples from a gaussian, the max is around 2.6 SDs. We have . With this estimate we still get around 40 IQ points.
As discussed above, similar gains could be gotten while still having a strongly genetically related father. For example, you could use more donors, and replace only the bottom-average-scoring half or quarter of the father's chromosomes.
This has a notable implication: If it's possible to nondestructively isolate chromosomes by index, then it's possible to make very intelligent children. For example, if using a centrifuge to separate many chromosomes by mass works to make bands of different chromosome index, and there's some way to isolate single ones (e.g. into liposomes), one could do this.
Selecting a diploid source of haploids
Suppose you extract SSCs from one or more men, apply chromosome selection to make a frankenSSC clone, and transplant it back into a man's testes. How much selection power do you get?
If we ignore problems with homozygous SSCs, you can do at least as well as simple haploid chromosome selection: you just double up on the best chromosome at each index. If there are several donors, you can use the top 2, which should fix homozygosity issues.
But there's a further question: can you do better?
You can't increase the expectation of the resulting haploids, of course. But crossovers between two copies of the same chromosome literally do nothing, so the all-top diploid produces haploids with no variance.
You can get greater variance by using some non-top chromosomes. Is this strictly a loss? No: crossover chromosomes can be higher scoring than the highest scoring single starting chromosome; see below.
So the plan would be to pick pairs of chromosomes (from multiple donors of course) that increase variance in the resulting haploids, while still being high average scoring. Then you'll do gamete selection somehow, or simply do embryo selection after fertilization with haploids from the frankenSSCs.
How do you pick which chromosomes to put in the SSC, to maximize the expected maximum embryo score from this procedure? What is the resulting expected maximum embryo score? I don't know. If you figure it out or run simulations let me know.
The power of recombinant chromosome selection
In recombinant chromosome selection, the input cell lines are not independent of each other. Rather, they are meiotic child cells of some set of donor cells. Examples:
- You start with one or more couples, and take several embryos from each.
- You start with several gametes from one person.
- You start with several people, and take several gametes from each.
- You start with one or more people, and use in vitro meiosis to produce several haploid cells from each person.
The advantage of recombinant over simple chromosome selection is that there is more variation in the scores of chromosomes that come from meiotic DNA recombination.
In terms of implementation, these methods are basically the same as simple chromosome selection. The main difference is that, in order to take advantage of the variance due to recombination, we have to sense multiple points on each chromosome. See the appendix "Cheap DNA segment sensing".
I haven't done the work to get a precise estimate of the power of recombinant chromosome selection in various forms. But see the following subsections. There are several complicating aspects:
- The distribution that meiotic recombination induces on the scores of the child chromosomes is quite importantly not a gaussian.
- The power depends to some extent how many (2, 3, or many) points on each chromosome we can distinguish; or in other words, how closely we can locate the crossover points.
- The power depends on how many crossover points occur. While the average for sperm is around 1.1, it varies by quite a bit. Depending on the numbers, you might be able to get a bump in power.
- Crossover points don't happen at random points.
- See the appendix "Best crossover".
One-donor recombinant chromosome selection
To simplify further, we can analyze the case where we assume (unrealistically):
- We select chromosomes from the gametes / haploids from just one person.
- Every chromosome has exactly 1 crossover point.
- The crossover point is uniformly distributed throughout the chromosome.
- Each chromosome is a sequence of many small gaussian samples, whose variances add up to the variance of the whole chromosome.
See the appendix "Best crossover" for some discussion of what the highest-scoring meiotic child chromosome will be. The upshot is that the very best chromosome with 1 crossover will be about 1.25 SDs above mean (in terms of SDs of that chromosome), and the very best with 2 crossovers will be about 1.75 SDs.
To make a more reasonable assumption than optimality, we can consider that we're able to label each chromosome at about 6 places. This gives about 1 SD of selection power for each chromosome; in other words, about twice as powerful as simple chromosome selection on two chromosomes. If we do this for a whole haploid genome, we get raw SDs, or about 24 IQ points after a round of embryo selection.
My guess is that this is actually an underestimate, because there are often two or three crossovers in a chromosome. A gold standard feasibility check would be to get single-cell genotyping data from a sperm sample and then compute the results of chromosome selection on that set of chromosomes using real PGSes.
Many-donor recombinant chromosome selection
Now consider multiple donors. If we did average-chromosome selection, we'd get some result. If instead we do recombinant chromosome selection, what do we get?
I think we can get a lower bound on selection power as follows. First we sample each donor's average chromosome score. Then we pick the highest average score. Then we get a good recombination of the two chromosomes. The recombination will be about 1 chromosome-SD higher-scoring than the average score. In other words, we maximize over the average-chromosomes of a given index; then we add 1. We might do better by comparing the best recombinations between donors, rather than just comparing the average scores (though obtaining the average score is far cheaper, unless we reliably get near the best recombination from one person's haploid sample).
In the case of one donor, average-chromosome selection gives +0, and then we add 1, giving the same as the above estimate.
In the case of 2 donors, we have as the corrected power at each chromosome. Then we pass that through each of 22 chromosomes, and add .7 with an embryo selection stage at the end. This gives 5.2 SDs of raw selection power. Someone spot-check the methodology here! The formula I'm using:
For 2 donors, and embryo selection on 10 embryos (but correcting down by because only selecting the other haploid), this gives
Which is rather a lot. That's +31 IQ points with one mother, two fathers, and sperm chromosome selection.
Double recombinant chromosome selection
If oocyte DNA is tough enough to handle the mechanical stress, and sperm DNA can be labeled well enough, and we can isolate chromosomes, then we could do recombinant chromosome selection on both gametes.
In a spherical cow model, that doubles the power. The situation is complicated by the fact that oocytes might be in quite limited supply. However, the space of crossovers is quite limited; it's a two-dimensional space (for the positions of two crossovers, as oocyte chromosomes often have), or one-dimensional if we're only optimizing over the 1-crossover chromosomes. With, say, 10 oocytes, we should get in the ballpark of the best chromosome. Since the above estimate is already conservative, a rough estimate of double haploid recombinant chromosome selection is simply double the power (but without much benefit from a round of embryo selection afterward): around 6.5 raw SDs, or around 40 IQ points.
I'll repeat that:
With one mother, one father, and recombinant chromosome selection, you get more than +40 IQ points.
Oocyte chromosome selection has the unfortunate effect of destroying many of the available oocytes. Since IVF embryos quite often don't make it all the way to birth (due to trouble fertilizing, morphological abnormalities, or normal miscarriage). This is likely too much of a cost for many mothers, but it would be technically feasible, assuming that the chromosome extraction method can work reliably and with single cells (which may be very difficult). IVO would remove this obstacle by making oocytes with different crossovers abundant.
Another way to avoid this obstacle, and give more than one chance to extract the chromosomes, is ESC chromosome selection. You grow the zygote into an early-stage embryo, say for just two weeks. Then you label the DNA somehow, and take a few cells from embryos with target chromosomes. From those cells you extract the target chromosomes. This shouldn't destroy many embryos. However, there's an additional challenge: identifying which chromosome is paternal and which is maternal. This may not be hard, e.g. with FISH targeted to alleles that only one of the parents has.
Fractional haploid donation
Another permutation of chromosome selection is this: instead of making a haploid by selecting all the highest-scoring chromosomes from any of several donors, you have a main haploid progenitor (presumably a member of a couple looking to have a baby), and then several external donors. The main haploid will have some especially low-scoring chromosomes, which will be replaced by the best chromosome from the external donors's haploid chromosomes (assuming that maximum is higher than that the main haploid's version). This would allow the main haploid progenitor to be more closely genetically related to their child than a grandparent would be, though still less genetically related than a parent.
This table shows the selection power of different numbers of donors and chromosomes replaced:

In row , column , this table shows the result of doing recombinant chromosome selection on both haploids separately, with replacements each. For one parent's haploid contribution, we sample donor haploid genomes. For each chromosome index, we consider the maximum recombined chromosome from the donors at that index, and look at its improvement over the parent's chromosome at that index. For the largest improvements, we replace the parent chromosome with the donor chromosome.
To get the values for this protocol applied to only one haploid, divide by 2 (and add .7 for a round of embryo selection).
Look at the rightmost column. With 23 donors, replacing just 3 chromosomes in each parent's haploid is nearly an additional 3 SDs above double recombinant chromosome selection (shown in the left column, under 0). At 9.6 raw SDs, this protocol would add 58 IQ points. Even further:

(There might be more noise in this table due to the lower sample size.)
Here's the code for simulating random genomes (modeled as subdivided gaussians): https://gist.github.com/tsvibt/89cec1b4fd7d54be04ba34a0059bcc4c
Here's the code for generating the specific data in this table: https://gist.github.com/tsvibt/3a3104d5e3530306c02ee7cbd2cf7ad7
Method: Iterated recombinant selection
Method:
- Take a population of cells.
- Create haploid recombinant child cells via meiosis.
- Combine haploids together to make diploids.
- Repeat 1—3; dovetail sequencing some of the cells and selectively amplifying some of them.
- At the end, apply some EC-making method to make a baby.
Because iterated recombinant selection involves meiosis, an epigenomically correct state can't be maintained. Besides the reprogramming done during meiosis itself, maternal and paternal segments of homologous chromosomes would end up in a single crossed-over chromosome; the result is epigenomically correct neither as a paternal nor as a maternal chromosome. Therefore iterated recombinant selection has to be followed by an EC-making method such as in vitro gametogenesis.
That said, iterated recombinant selection is extremely powerful. Because it can progressively interleave smaller and smaller segments of the starting chromosomes, the achievable selection power is, in practice, unbounded. In other words, you could get so many raw SDs of selection power that it would literally be unsafe to apply them all to known traits. See the bullet point "Traits outside the regime of adaptedness" in the perils list.
In practice, the question about iterated recombinant selection is about the costs. My guess, based on preliminary analysis and simulations, is that you can get quite large effects—over 10 raw SDs—for a few hundred thousand dollars near the beginning, or less. (The costs should then follow an innovation curve.) See the appendix "The costs of iterated meiotic selection" for more detail.
Iterated embryo selection
See "History of Iterated Embryo Selection", Branwen 2019[96].
In iterated embryo selection, the haploid cells are created via in vitro spermatogenesis and in vitro oogenesis. The resulting gametes are used to create embryos, which are grown in vitro; then the cycle repeats.
Iterated meiotic selection
The first description of IMS I'm aware of is "Meiosis is all you need", Metacelcus, 2022[97]. IMS is roughly the same as iterated embryo selection, except that instead of trying to recapitulate the whole reproductive cycle including spermatogenesis, oogenesis, fertilization, and embryonic growth, you directly induce meiosis in stem cells. Instead of fertilization, you simply fuse two cells through electrofusion[98]. An illustration:
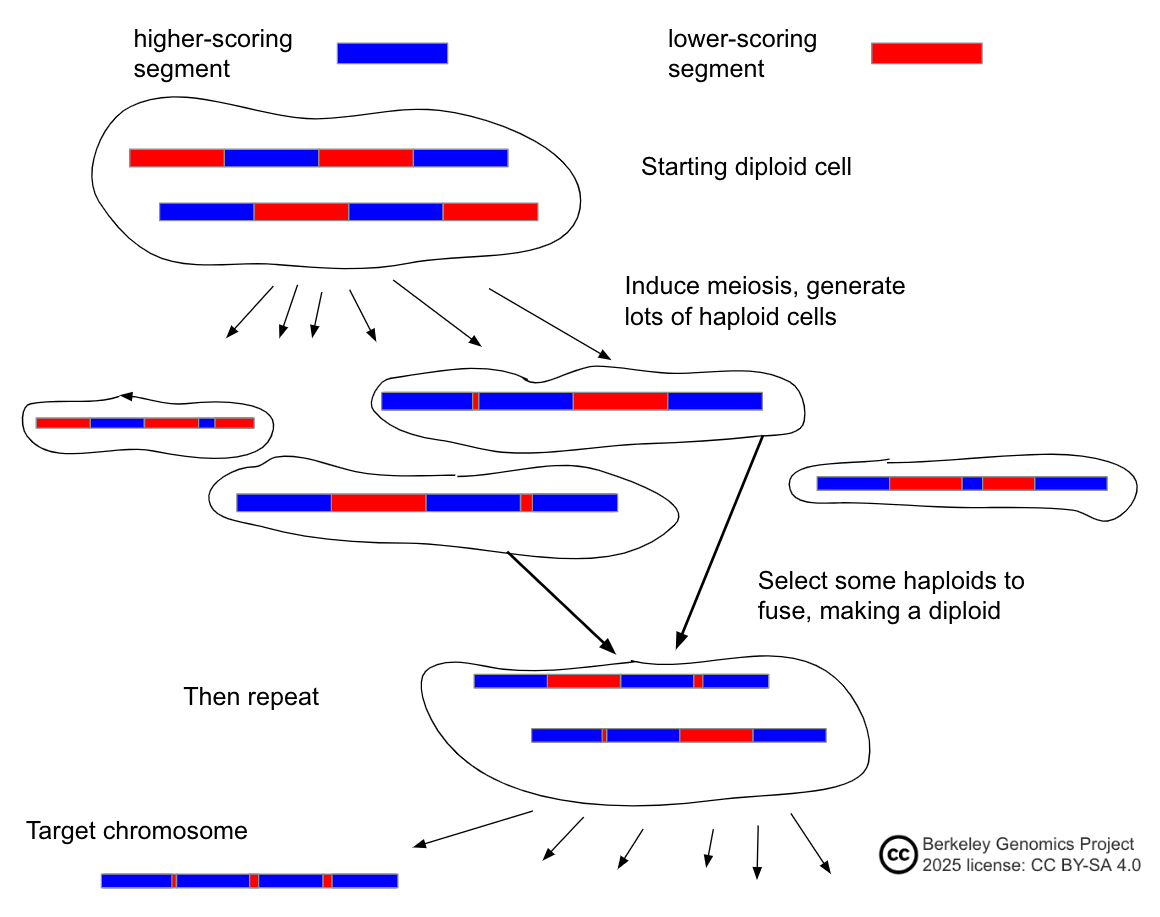
As Metacelsus points out, there are three major advantages to IMS over iterated embryo selection:
- It's more powerful to select haploids rather than diploids.
- I'm not sure exactly how much, because the situation is complex, but a very rough rule of thumb is that haploid selection might go about times faster than diploid selection. (This is based on the simple case of selecting from a gaussian vs. selecting from each of two i.i.d. each with half the variance of .)
- Meiosis is much faster than gametogenesis. In humans:
- Spermatogenesis: very roughly 3 months.
- Oogenesis: at least 6 months naturally; could maybe do it faster in vitro.
- Meiosis: 3 weeks, maybe possible to do it in 2. That's roughly an order of magnitude faster.
- Someone who is good at clocks please help me budget this.
- IMS doesn't require IVO and IVS.
- Gametogenesis involves both meiosis, and also epigenomic reprogramming and other morphological changes. Recapitulating the whole process may be significantly more complex than merely causing a cell to undergo meiosis. IMS only calls for inducing meiosis, which may be simpler to discover methods to do.
- However, IMS does still require some form of EC-making at the end (which could e.g. be IVO).
A fourth advantage over iterated embryo selection: IMS does not involve the creation of embryos. Haploid cells that haven't been epigenomically corrected are not functional as gametes, and fusing them to form a diploid stem cell does not create a cell that could naturally grow into a embryo. This should alleviate some life-protecting concerns about killing many embryos in vitro, which would happen in iterated embryo selection.
The main challenge to implementing IMS is to induce cells to undergo meiosis in vitro. This has not been achieved scalably and with appreciable efficiency in human cells. See the discussion of meiosis in the earlier section "IVG as an epigenomic correction method".
In the human body, meiosis only happens in the germline—spermatocytes and oocytes. So it might be that the easiest way to get meiosis is to invent in vitro gametogenesis. On the other hand, the bar is lower for meiosis compared to gametogenesis; no EC-making is needed, for purposes of IMS. Natural gametogenesis takes a long time because there are several other processes happening (epigenomic reprogramming, oocyte growth), and because gametogenesis is tied to the whole organism's development. So it's plausible that inducing meiosis is easier than full IVG, and could be applied for much faster meiosis. As a suggestive observation: during the growth of oocytes in primordial follicles, part of the function of cumulus granulosa cells is to prevent the oocyte from resuming meiosis[65:1]. Maybe removing cumulus cells when imprinting has been established, even if the oocyte isn't fully grown, would speed up in vitro meiosis compared to natural oogenesis.
One obstacle to full IMS is the culturability of the haploids. To do selection over haploids, you'd have to proliferate them monoclonally so you can take a sample and destructively genotype them, and then selectively amplify some clones. Culturability may or may not come for free. Some strange sort of human haploid embryonic stem cells have been cultured[99]. (Even without culturability, the cycle speedup is a massive benefit; I'd guess that matters more than haploid selection. Also, low-fidelity non-destructive DNA sensing would allow one to select at the haploid level quickly and without culturing the haploids; see the appendix "Cheap DNA segment sensing".)
Another possible obstacle is that induced meiosis might be error-prone. Natural gametogenesis likely involves upregulating DNA protection and repair mechanisms, and involves culling meiotic cells with damaged DNA. In vitro meiosis might lack some of those mechanisms by default, so it might produce haploids with more DNA damage. Damaged cells can be filtered out iteratively, but this adds sand in the gears.
As I said above, with IMS the question of selection power is less "how much selection power", and more "how expensive for how much selection power". See the appendix "The costs of iterated meiotic selection" for more detail.
Auxiliary: Enhancements to meiotic methods
There are two hypothetical methods that would specifically enhance the power of methods that involve meiosis, such as iterated meiotic selection or IVG with gamete selection. These methods don't matter too much, as the bottlenecks are elsewhere, but I think they're interesting conceptually.
The first is hypercrossover. You somehow influence the diploid cell as it meioses, to make it have several crossovers per chromosome rather than the typical 1—3.
Segments would move around more freely, thus making themselves available to be selected-for more independently and therefore rapidly, e.g. in iterated meiotic selection.
Hypercrossover would also, in theory, increase the ceiling for single-round meiosis. If you made very many gametes (billions?), you'd do noticeably better with hypercrossover than without. (Maybe with only thousands; someone could check.)
Finally, hypercrossover would be genuinely powerful combined with a round of chromosome selection. The highest-scoring chromosome would be higher-scoring by some multiple, like 2x or more, compared to the highest-scoring chromosome with only 1 crossover. (As a touchpoint, consider that with 3 crossovers, you're doing something like 4-segment selection, which is times as powerful as simple chromosome selection.)
Crossover rates can be increased in mice by knocking out a crossover-limiting gene[100]. I think the increase is about 30% here, based on the centimorgan numbers. This method would be bad for several reasons, and in general hypercrossover might cause DNA damage; but gentler methods, combined, could hypothetically produce several crossovers per chromosome.
The second auxiliary method is targeted crossover. You somehow influence the location of crossovers. If the influence is strong, then e.g. IMS becomes much cheaper, as you don't need to produce nearly as many haploids waiting for your desired crossover. If you can influence all the chromosomes at once in an in vitro gametogenesis protocol, you could have a vastly cheaper implementation of optimal gamete selection.
I don't know if targeted crossover is plausibly feasible. A possible method would be targeted double-strand breaks in diploid cells. You have two homologous chromosomes and , e.g. the two chromosomes of index 1. You have a desired crossover point. So you cleave both chromosomes at the crossover point using CRISPR-Cas9. Then you sit and hope that the cell's homology directed repair mechanisms swap the homologous arms, effectively making a crossover. (You'd still need some way of getting the resulting recombined chromosome into a cell to use as a gamete or zygote, e.g. via chromosome selection.) An illustration:
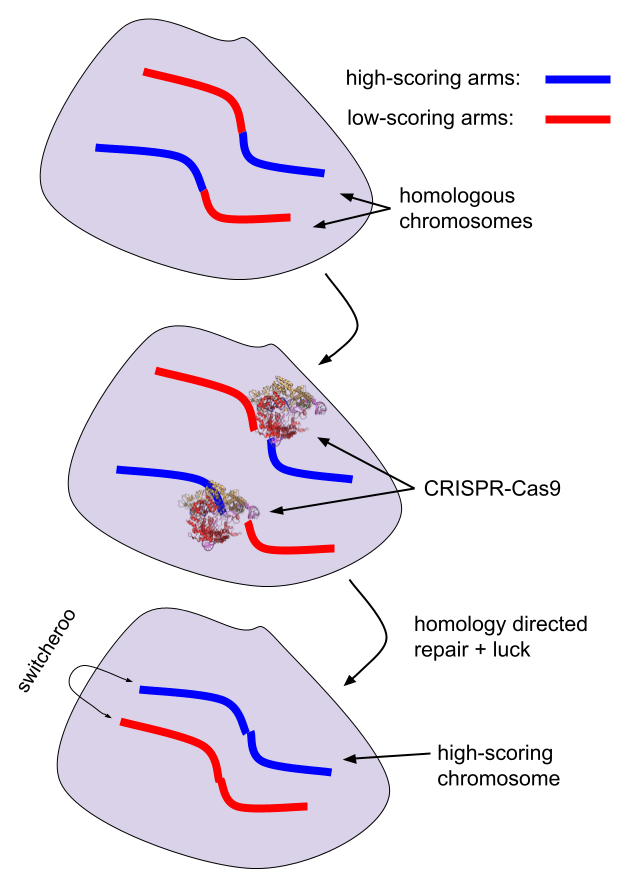
[(The protein pictured is Cas9, visualization from Synpath.)]
Method: Iterated multiplex CRISPR editing
See GeneSmith and kman (2025) for the source of the estimates of the strength of editing[2:1]. These estimates haven't been peer reviewed. They are based on a PGS that correlates at 0.35 with IQ, so they aren't directly comparable to estimates for selection methods given in this article, which assume 0.4.
Method:
- Apply a set of CRISPR editors (e.g. base or prime editors) to some stem cells.
- Culture the cells, perhaps monoclonally; if possible, sequence them to check for on-target edits made and off-target damage done.
- Repeat 1 and 2, correcting errors if possible.
- Make a baby from the edited cell.
(Aside: It would also be possible to safely edit an embryo directly. You'd use prime or base editors to edit the zygote (1-cell embryo), and you'd apply some treatment that deactivates the editors after a couple days. Later you sequence the embryo to ensure that there isn't any dangerous DNA damage. Since you edit at the 1-cell stage and then deactivate the editors, there shouldn't be mosaicism, so you can be fairly confident that the lack of damage in cells you biopsy and sequence really does mean that the embryo itself is fine. However, the restriction to only edit at the 1-cell stage means you can only do 1 round (perhaps multiplexed, like 10 or 20 edits). So this method isn't a strong GV method.)
DNA damage from editing
A general issue is dealing with DNA damage caused by editors. There are at least four kinds of damage:
- Chromosome translocation/fragmentation. Some editors, such as the original Cas9 system, cause double-strand breaks in the DNA. Usually double-strand breaks are repaired by reattaching the two ends of the chromosome that was broken. But sometimes the ends aren't repaired, or are repaired by stitching together two non-homologous broken chromosomes.
- Off-target edits. All CRISPR editors have some amount of mistargeting, where they simply edit some locus other than the intended target. The guide RNA that makes the editor target some DNA segment can also sometimes bind to other similar DNA segments at different sites in the genome.
- Indels. At the editing site, when an editor breaks/removes/unzips some DNA, unwanted nucleotides sometimes attach to the exposed DNA ends, causing unwanted insertions. Editors can also cause unwanted deletions.
- Bystander edits. Base editors cause bystander edits. After binding to the target site, they convert the target base to another nucleotide. But they sometimes also convert nearby bases of the same type as the target.
Depending on the rates of errors, this damage can probably be greatly attenuated by constantly sequencing the cells and discarding ones with any / many errors. They can also potentially be fixed by editing again (though this adds complexity and potentially significant delay, as you'd have to reengineer a new guide RNA to make the unexpected edit). Base and prime editors have much lower error rates than older double-strand breaking CRISPR systems, but still cause damage. DNA damage from editors might be especially bad compared to natural mutation, given that it introduces probably very rare de novo mutations.
Naive ESC editing
As discussed above in "Using natural reproductive DNA", it may be possible to maintain an ESC in a naive state and edit it, and then use that cell as a zygote. There will likely be issues with maintaining the epigenomic state, especially the imprinting. As with any method that tries to operate with naive ESCs to be used directly as zygotes, this might be a progressive problem: we might see significant early achievable gains, but then pushing forward another couple iterations (requiring several passages and proliferation) might continually degrade the epigenomic state.
Hulk sperm
Another EC bypass method, named by GeneSmith, is as follows.
- Biopsy the basement membrane of a testicle to obtain gonadal tissue, including spermatogonia.
- Edit the spermatogonia in vitro get target edits.
- Make a monoclone and verify the integrity of the editing spermatogonia.
- Edit in a fluorescent reporter near a gene that's only expressed in sperm.
- Re-transplant the edited spermatogonia back into the testicle.
- Wait a few months.
- Obtain a sperm sample.
- Isolate the magic glowing green sperm, e.g. using FACS.
One minor issue is that, if I understand correctly, the fluorescent reporter is a whole gene knock-in, so it would be inserted via normal double-strand-break CRISPR-Cas9 editing. Double-strand-break editing is prone to DNA damage such as indels around the break site. One always verifies the embryos at the final stage; so hopefully this just means that some smallish fraction of embryos will be unsafe to implant.
A larger issue is maintaining the spermatogonia in their support niche. If they fall out of their niche, they might not be able to proliferate in general, or differentiate into sperm, or have all the right epigenomic imprints.
An alternative method would use iPSCs instead of extracted spermatogonia. Then, after editing, you'd convert the iPSCs to spermatogonial stem cells, and then transplant them.
As mentioned in the earlier section "Features of GV and EC methods that affect compatibility", an issue with spermatogonial transplant into testicles is that GV (e.g. edited) spermatogonia are likely to be stressed and imperfectly maintained, and therefore are likely to be outcompeted by normal spermatogonia and die out. A possible workaround is to just transplant a large amount of GVed SSCs into the testes. I think that FACS can be quite sensitive, so if even a few glowing sperm make it out, they may be retrievable.
Magic rainbow sperm
Here's a weirder kind of editing: in vivo editing. This is known as the "we zap your balls until you make magic rainbow sperm", which is not a good method. How it works:
- Transfect a CRISPR editing package into testes in vivo. The package contains instructions for several desired target edits (that is, several guide RNAs), and also a fluorescent reporter protein to be expressed only in sperm.
- Zap the testicles to electroporate lots of cells, including hopefully many spermatogonia.
- Repeat 1 and 2 using different colors for each round, so you'll be able to tell which ones got edits from many different rounds.
- Wait a few months.
- Obtain a sperm sample.
- Isolate the magic glowing rainbow sperm, e.g. using FACS to find the brightest and most colorful sperm.
This isn't the best method: the edited spermatogonia would be stressed, knocking in several fluorescent proteins might be an issue, the editing would be inefficient because it wouldn't reach many cells, and surrounding tissue might be damaged by the editing and the electroporation. But it is the funniest method.
Conclusion
See the Takeaways.
Some other random small things you could do that might be fun:
- Come up with more feasible ways of doing GV and EC.
- Make a dashboard for combining GV methods in complex ways and showing the resulting GV power.
- Make an IMS simulator / game. Use AI or something to come up with clever ways of scheduling meiosis, sequencing, culturing, and cell fusion, to get large effects at low cost. Try adding in hypercrossover, targeted recombination, or chromosome transplantation.
- Review / replicate / fact check this article.
- Compute some GV power numbers for making multiple children for one couple, taking into account the goal of having children with genetic overlap that's not much more than the overlap of siblings.
- Simulate hypercrossover. E.g., in practice how does the distribution of haploid meiotic daughter genomes change with different numbers of crossovers? How does the in-practice maximum chromosome score change?
My work is not being supported and I am approximately out of money. If you want me to continue working on what I think I should be working on, you can support my work by donating here. (I'll edit this message if I receive enough to last 1.5 years.)
Appendix: In vitro spermatogenesis studies
Here are some of the main studies:
- Some early studies include Tesarik et al. (1999)[101], Cremades et al. (1999)[102], and Tanaka et al. (2003)[103]. Notably, Tesarik derived two apparently healthy live human births from round spermatids produced from primary spermatocytes in vitro. However, another embryo implantation attempt resulted in an ectopic pregnancy, which is really bad—the embryo starts growing somewhere other than the uterus. Tanaka et al. got ok efficiency: around 5% spermatids from primary spermatocytes, on a culture of Vero cells (iPSCs from an African green monkey). These early studies had poor verification of haploid genome content, and very poor measurement of the epigenomic state of their in vitro-derived haploids. Tanaka et al. (2009) continued applying their Vero cell culture method, and were able to mature round spermatids into elongated spermatids and even some spermatozoa[104].
- Easley et al. (2012) cultured human iPSCs with mouse SSCs (spermatogonial stem cells). They found that about 40% became spermatogonia-like (according to a few markers), and that about 3.9% of them became haploid, some with other round spermatid-like characteristics. However, they didn't do scRNA-seq or methylation sequencing (because at the time, that technology hadn't yet been developed), so we don't know what their haploids look like epigenomically; they checked just 2 imprinting sites[105].
- Perrard et al. (2016) used a bioreactor to culture adult human seminiferous tubule tissue. SSCs eventually produced some spermatids and some mature spermatozoa. They didn't evaluate the epigenomic state of their spermatozoon-like cells[106].
- de Michele et al. (2018) took fragments of testes from prepubertal children who had died and cultured them for several months, finding that after a couple weeks some of the SSCs in the starting tissue had produced round spermatid-like cells, though again they were not well-characterized epigenomically[107].
- Sun et al. (2018) cultured human SSCs with human Sertoli cells filtered from adult testicular tissue, in a gel to make 3D tissue. They say the SSCs differentiated into round spermatids at a rate of up to 17.9%. They also did RNA sequencing and bisulfite methylation sequencing, showing significant similarity between their round spermatids and natural ones, though the similarity was far from perfect and I don't understand the results[108]. (They also showed that their human round spermatids could fertilize mouse oocytes.)
- Yuan et al. (2020) cultured human fetal gonadal ridge tissue, which probably contained prospermatogonia, and found that after many weeks there were around 5% round spermatid-like cells. In a first, they used single-cell whole genome sequencing to confirm that these haploids had proper DNA content from meiosis (the right number of chromosomes without major gain or loss). The spermatid-like cells could fertilize oocytes, though only one of 16 made it to the blastocyst stage (about a week into development). They only checked a couple imprinting regions, so we don't know to what extent epigenetic correction occurred[50:1].
- Hwang et al. (2020) turned human PGCLCs into prospermatogonia-like cells by culturing them with mouse testicular cells. They evaluated their cells with scRNA-seq, compared to natural prospermatogonia of a comparable stage, and found that they were broadly similar but with notable differences. I don't know how to evaluate the differences, but they do note that their prospermatogonia-like cells are enriched for some gene expression that's associated with infertility[51:2].
Appendix: Cheap DNA segment sensing
The problem of cheap DNA sensing
Suppose you have some cells, such as sperm or embryos, and you want to find out roughly what their genomes are, very cheaply and maybe without destroying them. If that's all you have, you might be in trouble. It's hard to detect much about a genome without some sort of DNA analysis that's destructive and expensive (SNP array genotyping, DNA sequencing), and that requires the somewhat labor-expensive step of isolating each cell individually.
But suppose further that the cells you have are descended from a cell with a known genome. E.g. you have sperm from a father whose whole genome you've sequenced, or you have stem cells created by combining a few different people's cells. Now the task is much more doable. In a sense, you already mostly know the genome of the cell you're interrogating! You just need to find out which large segments from the parent cell(s) made it into this cell.
Basic chromosome sensing
As a toy example, suppose you take two cells from two different people, and then create a single cell by randomly picking a full complement of chromosomes from the two given cells. (For example, you electrofuse the cells, and then wait for the tetraploidy to spontaneously correct through mitosis.) So, some process randomly picks two of the four given chromosomes of index 1; two of the four chromosomes of index 2; and so on for each index up to 23. Now, how to figure out the genome of this new cell? You mostly already know it. You just need to figure out which 2 of the 4 chromosomes at each index made it into the cell.
You could do this by sequencing or genotyping, though that would be relatively slow and a little expensive, as well as destructive. But it's also overkill. All you really need is bits of information per chromosome. In general, this seems to me like it ought to be doable, though I don't know of a method that would definitely work.
A method that might work and be non-destructive and fast, is FISH (fluorescence in situ hybridization). FISH puts fluorescent molecules into cells. The molecules are bound to DNA probes, which are targeted to bind only to specific shortish DNA sequences. They hybridize with the DNA, sticking to it for a while. Then you can literally see which chromosomes were bound or not.
So what we'll do is, for the four chromosomes of index 1 that we started with, we'll analyze their sequences to find one (or maybe a couple) identifying variants. E.g. maybe one of the chromosomes has SNPs , another has , another has , and the last has . If we label with an orange fluorescent protein, and label with a green one, then based on orange/not-orange and green/not-green, we can tell which chromosomes are in our cell! E.g. a cell that's just green is .
Sensing crossovers
Now a more complicated case: meiotic child cells. Natural meiosis—dividing from a diploid (46 chromosome) cell into a haploid (23 chromosome) cell—occurs as part of oogenesis and spermatogenesis; artificial in vitro meiosis might soon become possible. Meiosis recombines DNA from each pair of two homologous chromosomes in the parent diploid cell, producing a single chromosome that is, for example, the first half of the first parent chromosome, followed by the second half of the second chromosome. In human gametogenesis, it's believed that there are generally very few crossovers per chromosome; on average, maybe 1.1 in sperm, and maybe 1.8 in oocytes (though with substantial variation between people and within one person's gametes)[92:1]. If you're looking at a gamete, and you know the parent's genome, all you need to know is roughly where the crossovers (probably 1—3) happened in each chromosome, and then you know almost all of the genome of that gamete.
How to detect crossover points? If you have some DNA probes, such as FISH, you can detect a few SNPs, spread across the chromosome, that differentiate between the possible parent chromosomes that the segment around that SNP could have come from. Say you have a sperm and you label 5 loci, dividing the chromosome into 6 segments; you label dad's paternal chromosome's SNPs orange, and dad's maternal chromosome's SNPs green. Then if you see a pattern like [green, green, green, orange, orange], you can be fairly confident that the first half is maternal, and the last third is paternal. (You don't know where the crossover in the 4th of 6 segments occurred; and you don't know for sure that there weren't more crossovers, e.g. one at the end in the middle of the 6th segment, or two inside of a single segment.)
Basically, you just label the DNA segments that are your targets, i.e. the ones you're trying to concentrate together in one cell as much as possible. Then you pick the cell with the most labels, i.e. the brightest cell.
An obstacle with sperm
There is a major issue with this protocol, though: I don't know of a method for labeling sperm DNA in a sequence-specific way. Sperm DNA is tightly condensed through protamination, and is therefore mostly inaccessible. Some sperm DNA is only histoned rather than protaminated, so it's possible that it could be labeled. But the chromatin is very tightly wound, so labeling molecules would have trouble getting to touch most of the DNA.
It's also possible that one could intervene to mildly decrease the degree of compaction, and then label the DNA when it's more accessible. For example, various treatments such as dithiothreitol are used to decondense sperm chromatin[94:1]. This probably causes some DNA fragmentation, but not at a very high rate. (In theory this starts to have some nontrivial effect on the broad-sense epigenomic state of the DNA, and therefore makes the case a bit less airtight that the labeled sperm should be exactly functionally equivalent to normal sperm. But probably it's perfectly fine, assuming that essentially the only function of protamination is for compaction (in order to swim), rather than silencing (which is anyway accomplished by methylations and the lack of much cellular machinery in the sperm).)
But, this might be fairly easy, or might be quite difficult, as far as I know.
Another issue is that the sperm chromosomes might be stuck together, e.g. by proteins or by being geometrically entangled.
Cheap sensing at scale with FACS
They key advantage of cheap DNA sensing would be that you can run assays at scale, e.g. to select from many thousands or millions of cells based on some DNA features. How do you select the brightest cell? Luckily there is existing technology that does something like this: fluorescence-activated cell sorting, or FACS, which sorts cells based on optical properties. I don't know enough to say confidently that one could sort in the necessary way, but my guess is you can. You could sort by brightness; or you could have multiple color labels and sort by activation of many colors. However, FACS might by default break up large DNA molecules.
Sensing chromosome index
In some contexts it may be possible to separate chromosomes by index without being able to tell anything else about the chromosomes.
One way to do this is simply to look with your eyes (classic!) through a microscope at the chromosomes, and see which ones look bigger or smaller. But look at the sizes of human chromosomes:

[(Figure 1.3 from Gallegos (2022)[109].)]
One might, for example, be able to visually distinguish chromosomes 1 and 2 from the other chromosomes. But good luck trying to eyeball the differences between 10, 11, and 12. If you can visually distinguish them well, you could even just guess. If you have two 2-way confusions (say, 21/22 and 1/2) and a 4-way confusion (9—12), you have to try about 16 times to get one correct one. Kinda expensive in embryos and labor but not that bad! (S. Eisenstat suggests: instead of guessing, do selection not to average chromosome score, but rather to average score of indistinguishable sets of chromosomes. E.g. you pick the highest-scoring donor to take all of chromosomes 9—12 from.) Though this may be infeasible because sperm chromosomes are quite compacted, so their length might not be visible.
Another possible method would be to lyse the cells and then centrifuge the sample, so that molecules separate by mass. Another hypothetical possibility would be pulsed-field gel electrophoresis. My gippities suggest perhaps Hoechst staining and flow cytometry. I don't know whether these or other methods would work, or would damage the DNA.
Appendix: Best crossover
Suppose you have a diploid cell, and you cause it to undergo meiosis. At index 1, the diploid cell's two homologous chromosomes will recombine, creating one or a couple crossover points. The meiotic child haploid cell will have a single chromosome at index 1, a cross between the two parent chromosomes.
Consider very many meiotic children created from one cell. The question is: what is the score of the top-scoring chromosome 1 out of all the crossed chromosome 1s?
What does recombination do to DNA?
Backing up, what actually happens to DNA in the two chromosomes during meiotic recombination? The whole process of meiosis is complicated, but here we just need to know how DNA moves around between chromosomes.
There are two kinds of changes to DNA during meiosis: gene conversion and crossover. In gene conversion, a small segment of one chromosome is used as a template to copy over to the other homologous chromosome, overwriting that segment. In crossover, the two homologous chromosome totally "switch tracks", and they end up having exchanged some fraction of a whole arm, from one end of the chromosome up to the crossover point.
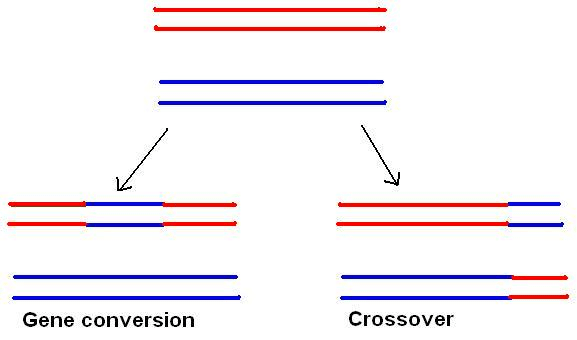
[(Diagram from Mikael Häggström, Wikimedia: https://commons.wikimedia.org/wiki/File:Conversion_and_crossover.jpg)]
In other words, a crossover of "A long time ago in a galaxy far, far away..." and "At the beginning of Elohim's creating of the skies and the land," could produce "At the beginning of Elohim's creating of a galaxy far, far away...".
How recombination actually happens is complicated:
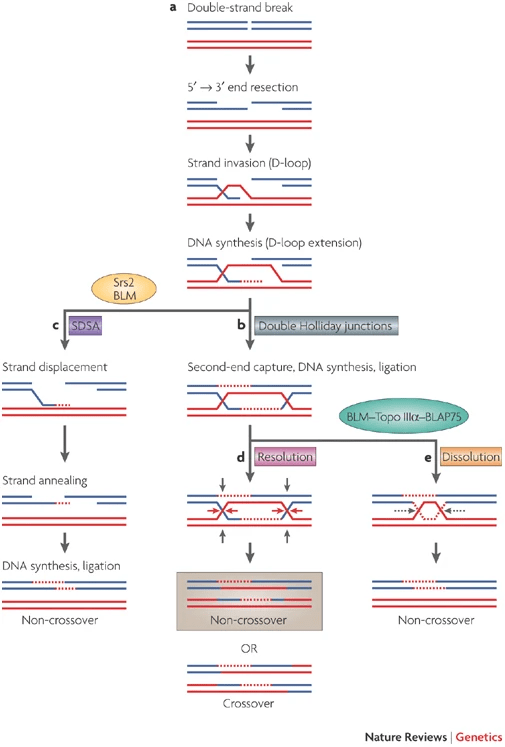
[(Figure 1 from Chen et al., 2007.[110])]
See also this beautiful animation by @WEHImovies (Drew Berry et al.), "DNA Break Repair by Homologous Recombination", which shows double Holliday junctions: https://www.youtube.com/watch?v=Xe-83tBcxhs
The main things we need to know:
- Gene conversion affects only a tiny portion of DNA, so we can largely ignore it.
- Sperm chromosomes have on average 1.1 crossovers or so, and oocytes are 1.8 or so, though both vary a lot between people and between gametes from the same person[92:2].
Math analysis of crossovers
As a modeling assumption, we can think of each chromosome as approximating a 1-D brownian motion starting at zero. The value of the brownian motion at, say, thirdway through the chromosome, is the score of the first third of the chromosome.
The difference between the local (e.g. SNP) scores of the two parent chromosomes also forms a brownian motion. As a simple base case, assume further that this brownian motion happens to end at 0. (This corresponds to the two chromosomes having exactly the same total score.)
Now the highest score among cross chromosomes will just be twice the maximum distance from 0 that this brownian bridge goes. (Plus the average score of the two chromosomes, which we can factor out of the analysis.) This problem has been treated by Kolmogorov and others; see e.g. "An excursion approach to maxima of the brownian bridge" for a source[111].
To illustrate what's happening, say we start with a brownian bridge:
[(This diagram is from Felix Xiao[112], and the following ones are modifications of it.)]
At position along the x-axis, we view the height of the brownian bridge as being the cumulative differences of the scores of the two chromosomes, up to along them. So e.g. if is 1/2, we see that one chromosome's first , i.e. the first half, is about 0.7 lower-scoring than the first half of the other chromosome.
To find the best crossover point, we just look at the place where the bridge deviates the most from zero:
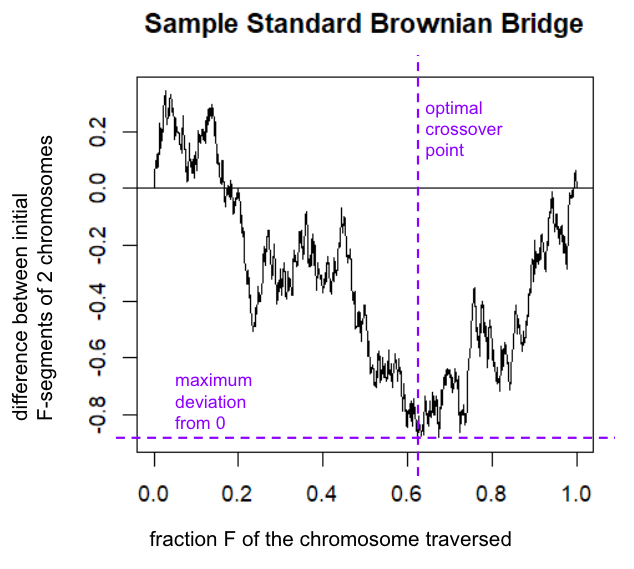
Then, if we get a crossover there, the resulting chromosome will have gotten the steepest difference in the first segment; followed by the steepest difference in the second segment, but with the sign inverted. ("Back in my day, we walked the whole length of the chromosome, uphill both ways.") The result looks like this:
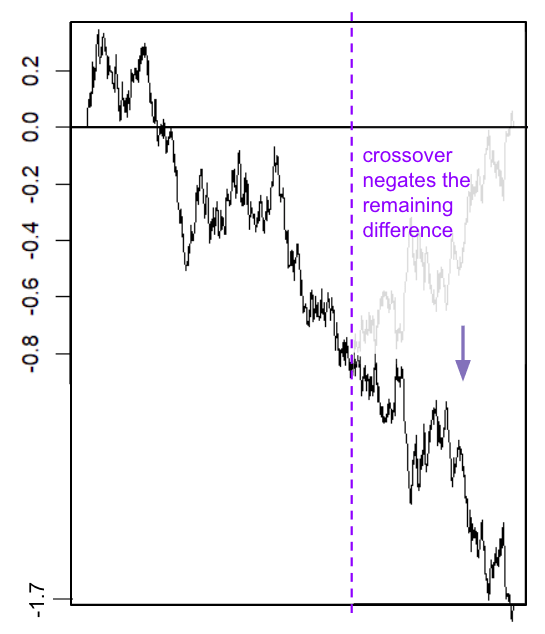
In this way, the crossed chromosome can have a score significantly far from zero, even if the parent chromosomes have the same average score.
For the full case, where the chromosomes can have different total scores, there are complications. E.g., we're uncertain about the endpoint of the bridge, and we're not just asking for the maximum distance to 0.
The case where there are 2 crossovers is additionally complicated.
Simulation results of crossovers
A simulation of recombination gives:
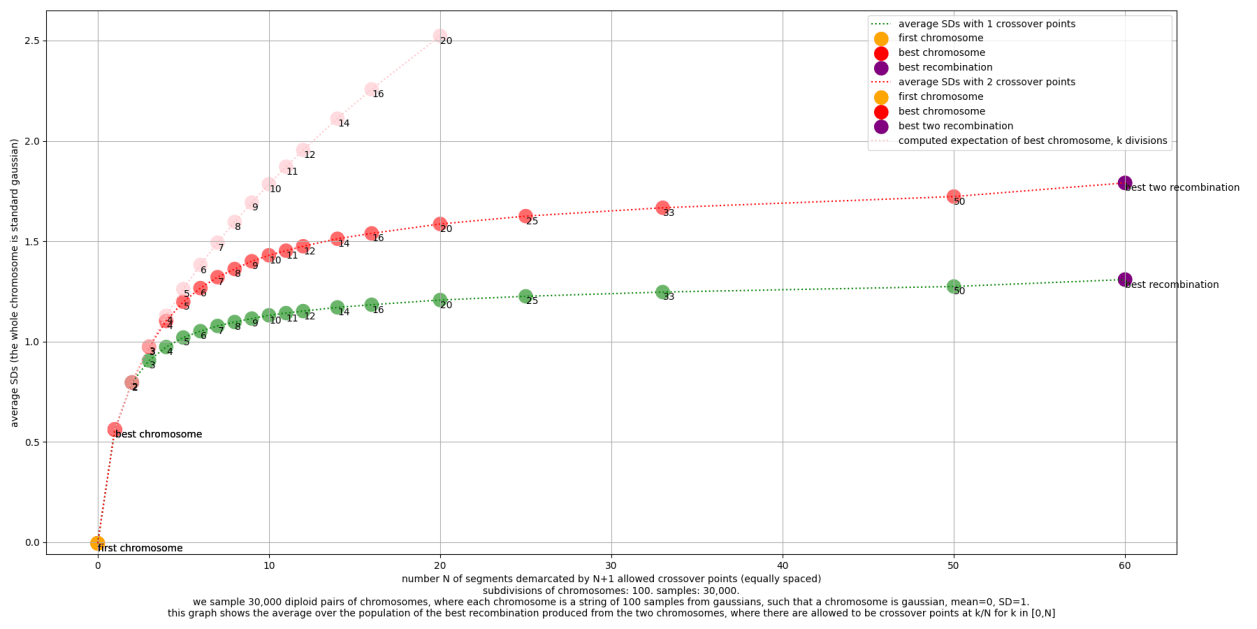
The simplest info in this graph is on the far right. It says that the best recombination with 1 crossover is about 1.3 SDs (in the scale of the whole chromosome), and the best recombination with 2 crossovers is about 1.8 SDs.
The rest of the graph shows the gain if you can only sense crossover locations with some precision. More precisely, the x-axis is the number of segments between allowed crossover points. At 1 on the x-axis, no crossover is allowed; at 2, only crossovers in the middle are allowed; at 3, crossovers at 1/3 and 2/3 are allowed; etc.
The pink line shows the results if you can freely mix and match the segments between chromosomes, however many crosses that takes.
We see that most of the benefit is gotten from 10 or even 5 segments. So sensing the chromosome of origin for 5 or 10 loci, spaced out through the chromosome, gets most of the benefit.
Here's the code for simulating random genomes: https://gist.github.com/tsvibt/89cec1b4fd7d54be04ba34a0059bcc4c. It computes maximum-score recombinations for each chromosome pair. By changing the number of segments per chromosome, you can simulate different levels of resolution used in recombinant chromosome selection. To replicate the numbers I give above for single crossover chromosomes, you can use something like this:
from shared_genome import *
def average_best_recombination(segment_count, sample_size):
chromosome_pairs = [DiploidChromosome(segment_count=segment_count) for _ in range(sample_size)]
return np.average([pair.max_recombination for pair in chromosome_pairs])
average_best_recombination(5, 10000)
E.g.:
>>> average_best_recombination(5, 10000)
1.0188024501193658
>>> average_best_recombination(5, 10000)
1.012057646519421
>>> average_best_recombination(10, 10000)
1.1343196475916841
>>> average_best_recombination(10, 10000)
1.1329019005532246
>>> average_best_recombination(30, 10000)
1.2360039835782262
>>> average_best_recombination(30, 10000)
1.2496364605062995
Refinements to the crossover model
The above analysis diverges from reality in several ways. Some of them:
Non-uniform crossover points
Crossovers don't happen at uniformly random points along the chromosome. Rather, they're somewhat concentrated in some areas, especially distal regions:
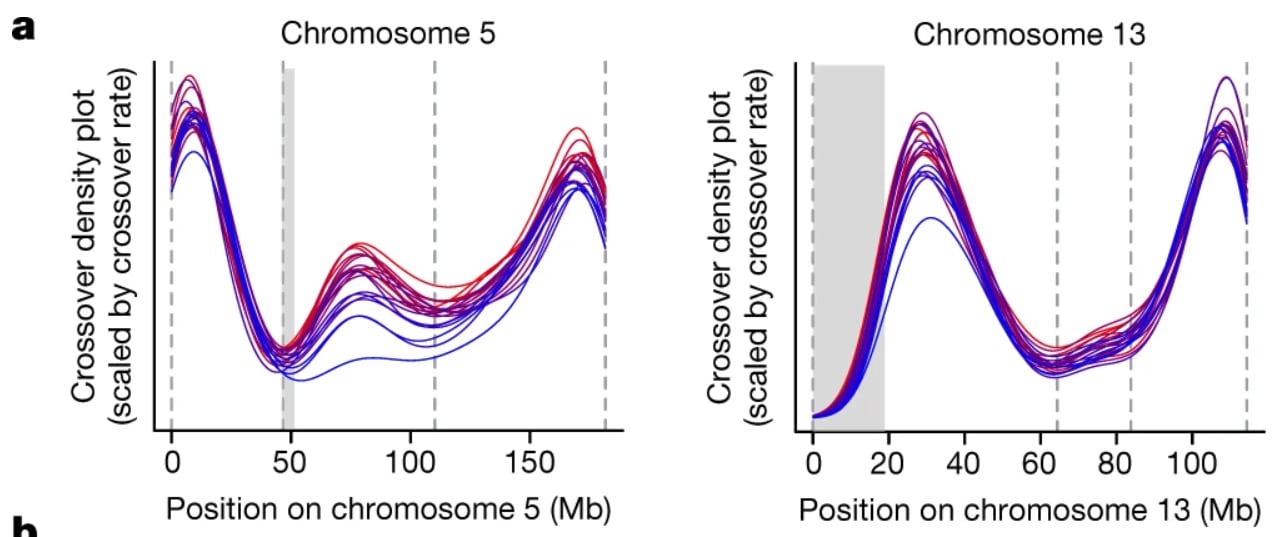
[(Figure 2a, from Bell et al., 2020[92:3].)]
This pattern at least qualitatively matches the distribution of functional DNA in the chromosomes; e.g. the ends and very middle of a chromosome don't do much coding or regulation (but rather have functions related to the physical structure of the whole chromosome). So if we rescale by relevance to germline engineering, the non-uniformity of crossover location would be somewhat attenuated (I don't know how much).
Further, for many purposes, a 5x difference in commonality of a crossover at a given point is not so bad. For example, for recombinant chromosome selection, even if the target crossover point is 5x less common, you just need to look at 5x as many chromosomes. Since you only had to look at tens of chromosomes anyway to get pretty close to optimal, this is not a problem for sperm chromosome selection. For oocyte chromosome selection, it would be somewhat of a problem, as even having 30 eggs would usually be considered an abundance. Then again, for ESC chromosome selection, if there are enough epigenomically intact cells (e.g. via twinning or just growth), no loss of oocytes / embryos is needed in order to attempt chromosome selection.
Multiple crossovers
The average number of crossovers per chromosome in sperm differs a bit between men, from 1 to 1.2 or so. However, the number per sperm varies much more: the standard deviation in the total number of crossovers in a sperm is greater than 4. That means chromosomes with 2 or more crossovers must be fairly common[92:4].
In the simulations above, the maximum score for chromosomes with 1 crossover is about 1.3 chromosome-SDs. But the maximum score for chromosomes with 2 crossovers is significantly larger, at about 1.8 chromosome-SDs; with 5 or 6 distinguished segments, the top-scoring 1-crossover is around 1 SD, but the top-scoring 2-crossover is around 1.3 SDs. There would also be some gain from selecting 3-crossovers. So multiple crossovers might significantly increase the selection power.
On the other hand, the relative positions of multiple crossovers aren't random. Rather, they tend to be somewhat nearby:
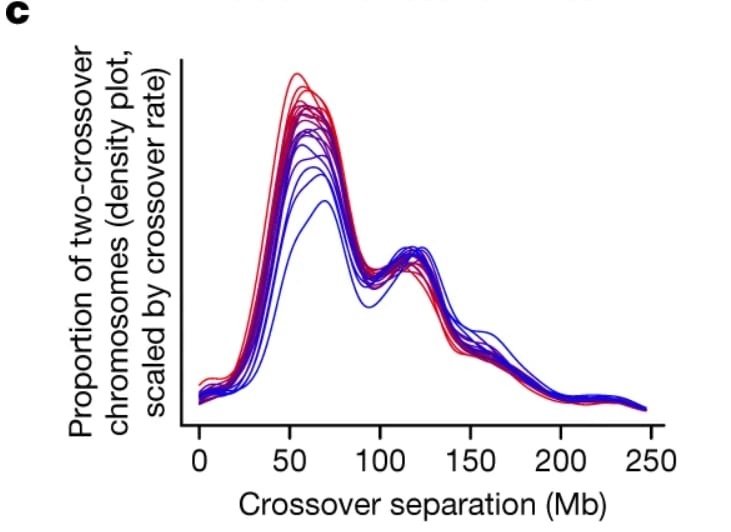
[(Figure 2c, from Bell et al., 2020.)]
This somewhat attenuates the effect of selecting 2-crossovers, though probably not all that much.
Empirically there are many 3-crossover chromosomes in sperm, though the rates vary a lot between men and between chromosome index:
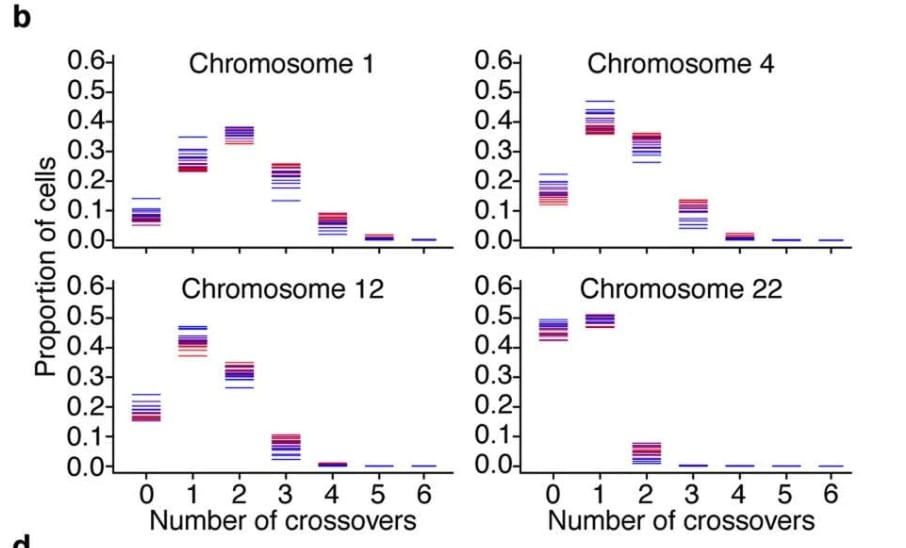
[(Figure 8b, from Bell et al., 2020. The bars indicate different donors; the colors indicate high or low average crossover rate for that donor.)]
Oocyte chromosomes have a much larger rate of crossovers than sperm—around 1.8 per chromosome, though again with a lot of variation.[113]
There is also non-crossover recombination, in the form of gene conversion. But this only transfers a tiny fraction of DNA between chromosomes—well under .01%[114]—and therefore doesn't much affect selection power.
Appendix: The costs of iterated meiotic selection
Some costs involved in IMS:
- Because you're trying to end up with a high-scoring cell, you want to maintain many positive variants in the population of cells. So you have lots of cell cultures at the same time, which takes money for equipment and culture media and labor.
- You have to do some form of DNA sensing to know something about the genomes of many cells, so you can select lines to propagate.
- You have to induce meiosis in very many cells. The process takes 2 or 3 weeks, and it will have some substantial attrition rate.
- You have to isolate and passage cells so you can proliferate lines as monoclones to sequence and select. (Well, not necessarily. You could do some more scalable process. E.g. you could use some fluorescent marker and FACS to find cells enriched in certain DNA segments.)
- All these processes can introduce mutations, e.g. cancer mutations, which you have to discard, adding more attrition.
Uncertainty in these costs makes it hard to estimate in advance the selection power per cost you can get with IMS. Aside from all these costs, the scheduling problem of when to combine, meiose, sequence, or discard which cells, is quite nontrivial; I don't understand it satisfactorily. It seems like it would be fun to build a proper simulator for the task, and then come up with and test strategies or do machine learning. Let me know if you're interested in doing that (it's not a priority for me).
The power of segmented selection
The simplest way to think about the power of IMS in general is to think of IMS as being "like chromosome selection, but for subdivisions of chromosomes". Instead of selecting the top-scoring chromosome 1, we select the top-scoring first half of chromosome 1 and the top-scoring second half of chromosome 1. We do this by arranging that the chromosomes with the appropriate crossovers (corresponding to our selection of segments between the crossover points) will make it into one cell together. An illustration:
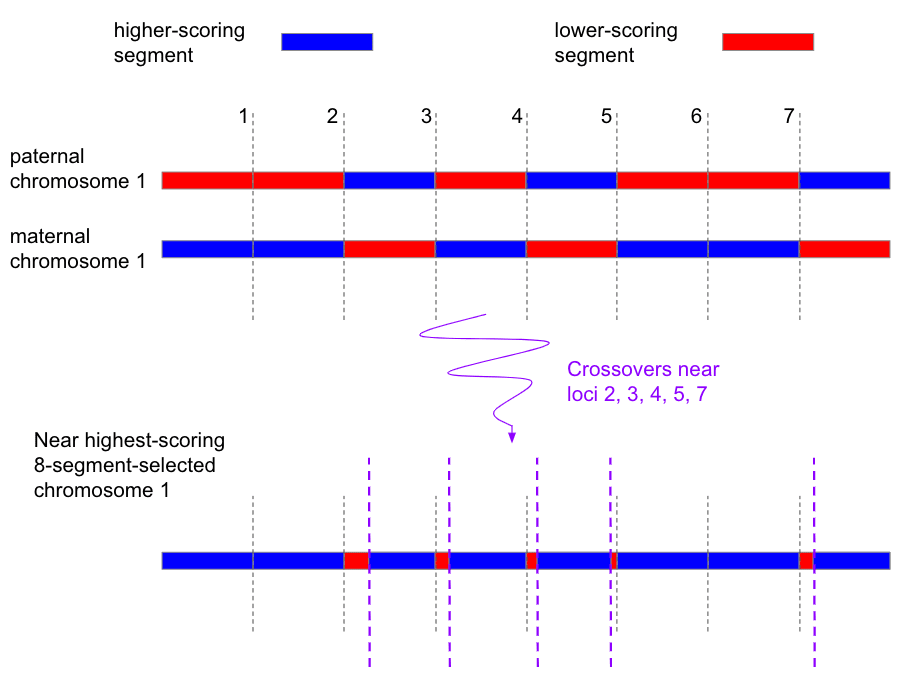
If you double the number of segments that you select but keep everything else the same, how much does selection power change? Each segment now has half the variance of the previous double-segments. So a segment SD is as big as an old double-segment SD. But there are twice as many, and the benefits sum. So the overall effect is that the selection power goes up by a factor of .
In other words, in general to compute the power of segmented selection, we can just multiply simple selection from a gaussian by , where is the number of (equal-length) segments.
For example, suppose we want to make a haploid cell from a single diploid. That means we'll get two choices for each segment; that means our base selection power is about 0.56 SDs.
If we just do chromosome selection, that's 23 subdivisions. The total variance of the haploid is 1/2 the total population variance. So the scale factor is . This says that chromosome selection should give SDs, which is roughly what we got for haploid chromosome selection above. (The divergence is because here we used 0.56 for best of 2 from a standard gaussian, and because we're allowing selection on the 23rd chromosome.)
Now suppose we do segmented selection with 92 subdivisions. In other words, we're dividing each chromosome into 4 equal segments, and then picking the top-scoring of each segment. How strong is this? Multiplying the segments by 4 will multiply the power by , which I believe is 2. That gives total raw SDs, or after embryo selection, or 27 IQ points.
Breakdown at high granularity
How about 10 subdivisions? Now we get raw SDs. After embryo selection, that's about IQ points. However, at this level of granularity, these mathematical estimates may start to significantly break down in reference to actual genomes. The issue is that with 10 subdivisions of 46 chromosomes, we're looking at around 500 DNA segments in a whole genome. A PGS might only know about dozens or hundreds of SNPs. So it doesn't make sense to directly model a segment as having a score sampled from a gaussian, as many segments will only have a few or even no effectful SNPs. On the other hand, the variance is still there somewhere, so one could imagine having segments with uneven length but evenly distributed variance; I'm not sure exactly where things really break down.
That said, in the case of IQ, the trait is highly polygenic—maybe on the order of 10,000 variants are involved—and a substantial fraction of the trait variance is explained by a PGS, involving over a thousand variants. Furthermore, the SNPs associated with IQ do tend to be spread out all across the genome, rather than being highly concentrated in smaller regions. See for example:
(This figure shows p values and doesn't show effect sizes, but the point stands.)
[(Figure 1a from Savage et al. 2018[115].)]
Finally, in practice, parents will want to select for several traits. Effectively, they'll select for a combined PGS, which may be a weighted sum or a more complicated function of PGSes for diseases, health, personality, IQ, or other traits. The combined PGS will have thousands of genetic variants to select for, spread across the whole genome. So these estimates of total raw SDs might mostly extend all the way to 10 segments per chromosome.
Segmented selection with one or more donors
If instead of making just one haploid, we make both, the gains add together, i.e. they are doubled: >50 IQ points for 4 subdivisions, or 80 IQ points for 10 subdivisions.
Say that instead of making a haploid from one parent, we make a haploid from two parents, to make a child with two genetic grandfathers and one genetic mother. That means that instead of sampling from 2 segments to get each 1 segment, we sample from 4 segments to get each 1. The expected maximum of 4 samples from a standard gaussian is about 1 SDs above the mean. Here's a snippet you can check these numbers with:
import numpy as np
def expected_max(test_runs=10_000_000, k=2):
X = np.random.randn(test_runs, k)
return np.mean(np.max(X, axis=1))
So with two parents to make a haploid, we get almost twice as much selection power as with one parent. For example, dividing each chromosome into 4 segments, we have 92 segments. The selection power is raw SDs. After embryo selection, that's about 55 IQ points. Doing this for both haploids is too many IQ points. Here's a table giving the numbers for segmented sampling of a standard gaussian, from here:
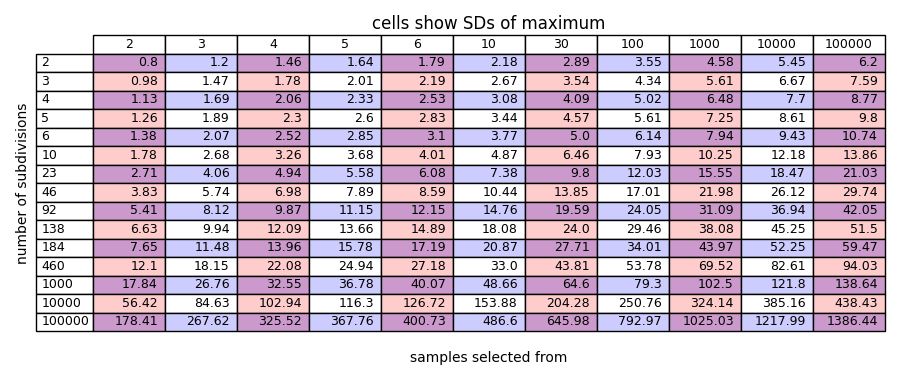
One way to interpret these numbers is that the column labels (2,3,4,5,6,10,30...) say the number of copies of each chromosome you start with, and the values given in the cells show the SDs of power you get from making both haploids separately given that many chromosomes. So the 2 column is the most normal case; it shows two parents who each make a haploid from their own genome (which gives 2 chromosomes of each type) with segmented selection, and then combine them. The 4 column shows the case with two parents to make each haploid. (The odd-number columns are not very relevant in this context.)
The row labels (...,23,46,92,...) show the number of divisions of the whole genome that are involved (including the separation between chromosomes). So the 46 row shows diploid chromosome selection. The row shows segmented selection with 4 subdivisions. As mentioned, a while past 184, things might start to break down.
Already with a normal, realistic case, IMS provides strong GV. In the 2 column (i.e. just one couple, two parents), and the 184 row showing 4 subdivisions, we get 7.6 raw SDs, or 46 IQ points.
The table can also be read as describing making one haploid; you just divide the values in the cells by .
The cost of poor man's chromosome selection
Keeping in mind that the optimal scheduling problem for IMS is complicated and hard to analyze, we can get still some sense for the problem. One angle of attack is to simplify way down like this: Assume that, instead of meiosis, the division process simply picks one of two whole chromosomes at random.
This is like the problem of whole cell fusion and tetraploidy reduction, but with diploids and haploids rather than tetraploids and diploids. If you think about the problem a bit, I think you'll see that it's not so trivial.
It's easy to make some progress quickly: You just select haploids with a larger number of the higher-scoring chromosomes and combine them. This does work well, but it usually fails to get all 23 target chromosomes into one cell. The issue is that pretty soon, all the top-scoring cells are top-scoring because they came from the same earlier top-scoring cell, so they have a lot of overlap in the target chromosomes that they have, and leave some target chromosomes extinct. Of course, you can do more clever things, such as preserving rare top-scoring chromosomes, and trying to make pairs that stand to make the most gain (have the most disoverlap of target chromosomes). These work, but it's not straightforward to mathematically analyze.
There is, however, a "speed limit" we can describe. What's the best possible pairing of haploids to make? You always want to take your top-scorer, and then pair it with another equally high-scoring haploid, but with maximal disoverlap so that you have the most upside (in other words, the most variance in the number of target chromosomes in the child haploids). Suppose that magically you can always do this. How fast do you go?
There's another parameter to specify, which determines how good the next top-scoring haploid will be: how many haploids do you get from each cell? As a simplifying assumption, suppose you get enough samples so that you will gain (i.e. one SD, ish) additional target chromosomes, above the mean, where is the number of non-target chromosomes left in your top-scoring haploid.
So at each stage, you do . This converges quite quickly: . So, not all that much of a speed limit after all, but it can give us a target speed to aim for.
The cost of single-chromosome IMS
As a different approach to analyzing IMS, we can simplify the problem by assuming that we have a method for chromosome selection. Now the problem is: If the genome is just a single chromosome, how do you cheaply get a high-scoring recombinant chromosome? If you have a good answer to that, you apply it 23 times separately, in parallel, for each chromosome. Then at the end, you use chromosome selection to bring those chromosomes together.
Since whole cell fusion is a kind of chromosome selection, we've sort of broken the whole IMS problem into two independent pieces. You're weaving together chromosomes; and then you're putting them together. In reality, you wouldn't do these separately, because it's more efficient to do everything simultaneously "in superposition", i.e. both weaving high-scoring chromosomes and shuffling those chromosomes together into the same cell. So the costs don't directly add up.
How to do single-chromosome IMS? The simplest case is this:
- There is one chromosome type.
- Each chromosome is a pair , where the are bits . The meaning of is "this chromosome has a non-target first half and a target second half".
- Our goal is to get .
- We start with a diploid .
- We can sample haploids through "meiosis", where there's always exactly one crossover:
- Randomly pick one of the two chromosomes.
- Take some initial segment of the chosen chromosome, either nothing, the first bit, or both bits.
- Fill in the remaining bits with the tail end of the non-chosen chromosome.
This case is easy to analyze. Half the time meiosis picks to start with, and a third of the time it picks the middle for the crossover point. So if you sample about 6 haploids, you get .
What about if we divide the chromosome into more segments? How do you assemble the target segments into a single chromosome? E.g., suppose you start with
and you want to make
How do you schedule pairings and meiosis to get the all-s chromosome?
...
...
(Pause for thinking.)
...
...
We need a crossover at each comma, i.e. 7 total. By default, if you accept random crossover locations, the expected number of total crossovers increases by one each generation. So we'd need 7 generations. This isn't hard: In generation you sample until you get a crossover at location , adding 1s to the initial segment one by one in order. Surely we can do better?
One strategy you can follow is to assemble sub-chromosomes in a binary-tree pattern. We build up islands of 1s which double in size and halve in number each round. An illustration, in the smaller case of 4 segments rather than 8:
To step through in the case with 8 segments: In the first round, you sample enough haploids that you get every crossover point. Now you have
In the next round, we pair the first two of these and we pair the last two. We sample from each haploid until we get the desired crossovers, like this:
And then once again until we get the middle crossover.
This method takes roughly 8 samples for each pairing; it takes 3 serial generations (rounds of meiosis); requires proliferating 4 diploids; and requires producing and genotyping on the order of haploids from diploids. If we did this 23 times and then did chromosome selection, we'd have to pay these costs 23 times (in parallel).
I would guess this strategy is basically optimal for the simple case we're dealing with.
However, in real life, there's a lot of utility being left on the table, potentially. All the assumptions we made are unrealistic, obscuring both obstacles and possible better strategies. E.g.:
- Sometimes meiosis produces more than one crossover in one chromosome. So potentially you could go faster.
- Chromosome crossovers don't just happen at your favorite 9 spots, they happen all over the place (though by no means uniformly distributed). This makes it more complicated to decide which haploids to keep; target segments could be broken up in whatever fraction. On the plus side, you might get especially helpful crossovers.
- We modeled the chromosome as being made of some equal-length segments with equal spread of scores between the two homologs. In real life, chromosome scores are more like a brownian motion (speaking imprecisely). They'll have lots of random little patches that are rather higher or lower scoring than usual. This means there's opportunity to get lots of especially good segments of various sizes, efficiently.
- We modeled a single chromosome. In real life there are 23 chromosomes. We could, to a significant extent, parallelize the individual chromosome-segment-selection processes within one cell. In other words, by causing a single diploid cell to undergo meiosis, we get 23 crossovers, one in each chromosome; it may be that several of them are useful.
- We assumed that we start with a single diploid cell, and want to produce a single haploid cell. In real life, we might start with cells from several different donors, and then produce a diploid or haploid. This is more complex, but the additional genetic variants raise the achievable ceiling.
- We assumed we genotyped all the haploids. Ideally we'd use a much cheaper DNA sensing method. But either way, it's possible to not genotype every haploid, and/or not wait for genotypes to be processed before proceeding to the next steps. This is sort of like branch prediction in a CPU. E.g. we could start inducing meiosis in a diploid before we've gotten back the sequencing results, and then later scrap the resulting haploids if we hear back about the diploid and decide it won't produce the haploids we want. Also, we could use multiple sensing methods, e.g. very coarse FACS to pick generally target-enriched candidate haploids to make monoclones from, and then do some cheap SNP array to learn more about those monoclones before pairing.
- In real life, we have to at least sometimes do whole genome sequencing to screen for de novo mutations.
- In real life there are many decisions to be made about which cell lines from earlier stages of the process to continue culturing, in order to preserve variation, and when to bring them back to reintroduce variants.
- We considered 8 segments, a power of 2. But 6 or 10 segments might be more favorable. The simple binary tree doesn't work in this case.
Appendix: Detailed estimation of embryo selection
Note: For most purposes, this section gives too much detail. To just understand the expected gains from simple embryo selection, it suffices to look at some gaussian and ask about the order statistics. But, I wanted to understand the error bars a little bit.
Here's the code for simulating random genomes (modeled as subdivided gaussians): https://gist.github.com/tsvibt/89cec1b4fd7d54be04ba34a0059bcc4c
This isn't all of the code used to make the graphs below, but it's the core model used, so if you want to check its correctness or play with it, that's the place to start.
Simple embryo selection simulations
In what follows: Chromosomes are modeled as being made of 100 segments, with each segment sampled from a gaussian with 1/100 of the chromosome's variance, which in turn is proportional to the chromosome's share of total genome length. Meiosis uniformly at random picks a point along the chromosome, and a parity (which chromosome first), and puts the result as the child chromosome; one crossover each.
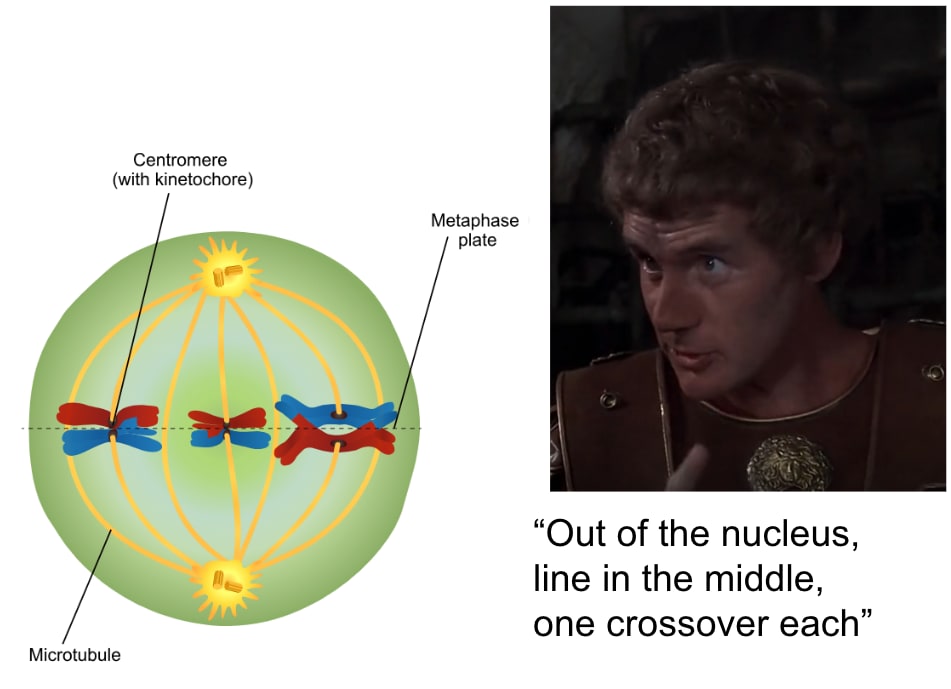
[(Modified from https://en.wikipedia.org/wiki/File:Meiosis_Stages.svg.)]
Analysis of simple embryo selection
We want to understand the effect of choosing the highest-scoring embryo that one couple produces. We can break this up into two questions:
- What is the distribution of embryos from one couple? In other words, what is the distribution of the score of embryos, sampled by combining two parent genomes randomly in the way that natural reproduction combines them?
- Given the distribution of embryos from one couple, what is the distribution of the highest scoring embryo out of a sample of embryos from the couple?
What is the distribution of embryos from one couple?
For each parent, for each chromosome, we sample a chromosome, independently of the other chromosomes. (That is an assumption about the biology—that crossover points and assortment aren't correlated across chromosomes.) Sampling a chromosome of index means sampling a recombination between the parent's two chromosomes of index . Modeling the distribution for one chromosome isn't trivial; see the appendix "Best crossover". But we can zoom out and just model the haploid as a sum of similarly-distributed variables. This gives approximately a gaussian, and then the embryo is just a sum of the two haploids, so it's another gaussian.
Now we just need to know the mean and variance of the gaussian. The mean is just the mean score of the two parents's genomes. What is the variance?
Heuristically speaking: The variance of all people is the same as the variance of all embryos. Embryos are sampled by first sampling a couple, and then sampling an embryo from that couple. The mean of the embryos from a couple is the mean of the couple's genomes. The score of an embryo from a couple is the mean of the couple, plus a sample from the couple's embryo-variance. So the overall variance of all embryos is the variance of the mean of a couple, plus the couple's embryo-variance. (This step needs more precision. It uses linearity of variance for the sum of independent variables. It also uses that conditioning on the couple's mean being some produces a distribution of embryos that's the same as conditioning on the mean being , but with means shifted up or down. The conditional distribution introduces small correlations between all the values of segments of chromosomes in both parents, but the point is that the shape of the conditional distribution (ignoring means) doesn't depend on the value being conditioned on; see this stackexchange answer.)
The couple's mean score is half the sum of their individual scores. Variance is additive, so . But halving the score will the variance (the score is in -space, the variance is in -space). So the variance of the couple's mean score is half the variance of the population. That leaves the other half of the overall variance of all embryos to be generated by a single couple's embryos. So a couple's embryo-variance is around half the population variance. (Again, this is not precise.) In other words, the typical couple's embryos are scores distributed as a gaussian with standard deviation roughly equal to the standard deviation of the population.
We can check this with simulations. Here's a graph plotting the variance of embryos from couples. (That is, the variance of total score, in the SD-scale of the population.) The green dots are couples. The x position of a point shows the mean of the couple's genomes, i.e. the mean of the couple's embryos. The y position of a point shows the variance of embryos sampled from the couple. The red line shows the averages of variances (binned together in some range on the x-axis). The gaussian is the probability density function (pdf) of a couple having a given mean score (with probability density on the y axis).
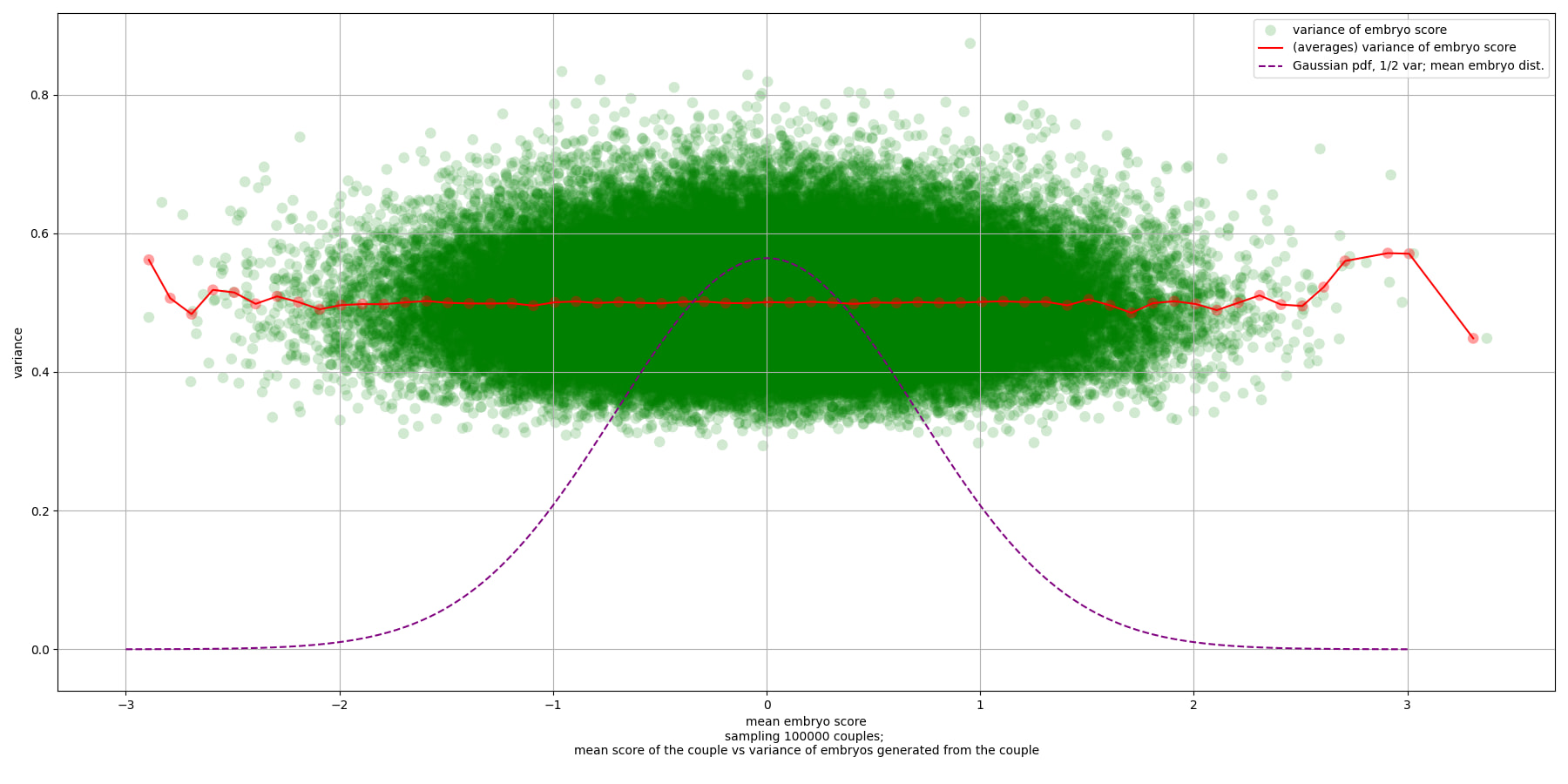
We see a few things:
- The variance is about 1/2.
- The variance does not change for couples far from the mean genome.
- (In theory, that is. In theory in theory, you never use up variance. In theory in practice, selecting very hard would use up variance—the genome isn't actually an infinitely divisible Weiner process, it's a finite list of base pairs. But in practice in practice, you don't use up much variance. E.g. if intelligence has on the order of relevant alleles, one SD is alleles. So you could select by 20 SDs, i.e. 1000 alleles, which is way too far anyway, and still have only used up less than half of the available variance. See the limits section of "The Power of Selection".)
- There is some variance in the variance of a couple's embryos. It looks like the distribution is a little skewed to be higher. It looks like the range is roughly . Taking square roots to translate to SD-space, we have roughly . So selection will work similarly well for everyone, but could have effects that vary by about 10%, compared to a modal couple with embryo variance .5.
For the sake of concreteness, below is a plot that shows seven couples. Each couple then generates many embryos, and we graph a histogram of the embryos's scores. For each couple we also graph a gray dotted gaussian with , centered around the couple's computed mean. Comparing the gaussian gives some sense of the variation in the variation; the couples are all quite close to their gaussians, but there's some noticeable differences.
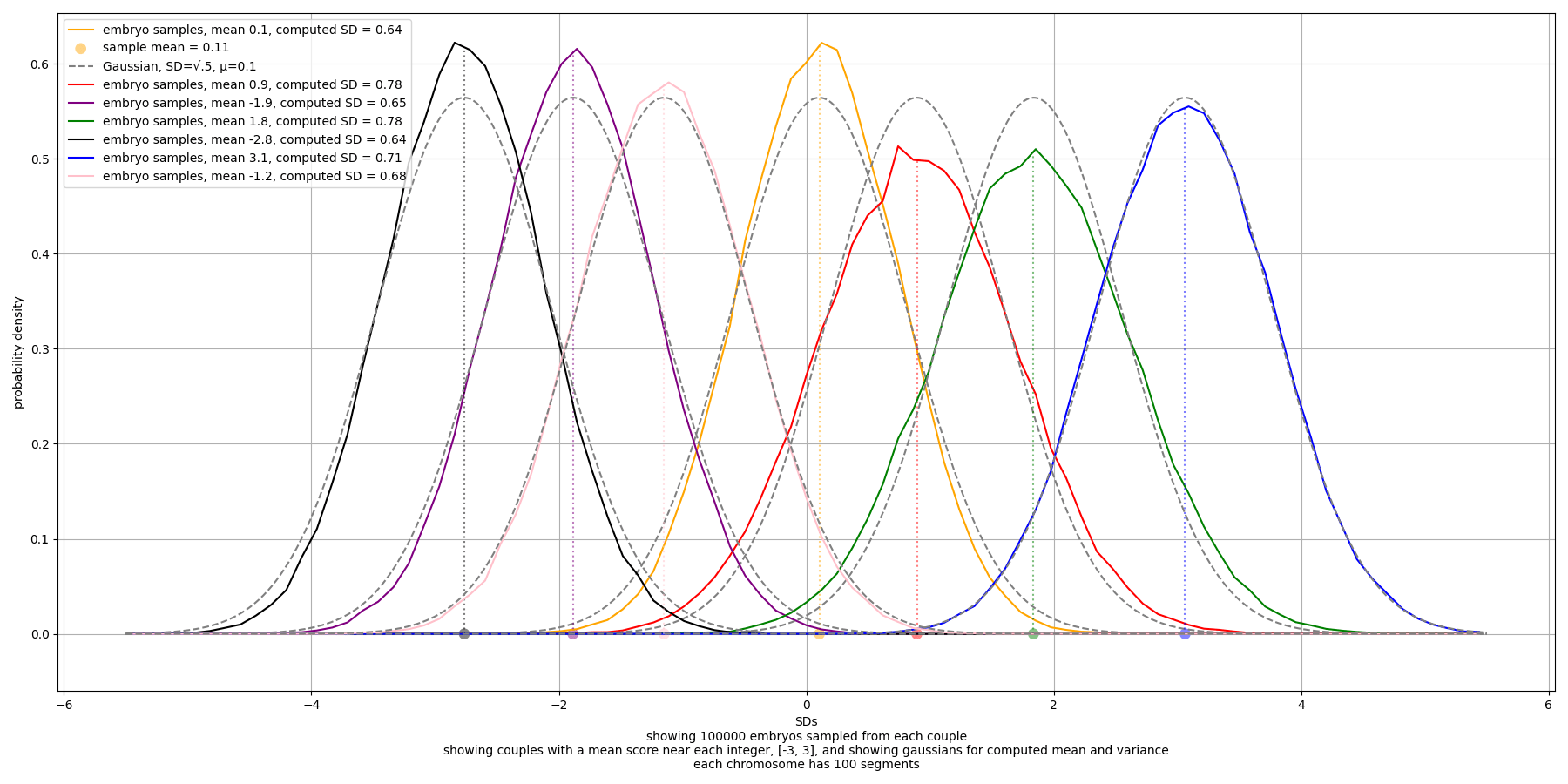
To get efficient estimates for sampling maximum embryos from couples, we want a better estimate of the parameters of the distribution of embryo-distributions. Below is a graph showing a histogram of, for each of 30,000 couples, the variance and standard deviation of embryos generated by that couple (computed by aggregating the couple's haploid variances). We see that the SD of the SD is about .045. The distribution is not a gaussian (e.g. because it has probability 1 of being nonnegative), but empirically it's pretty close to one.
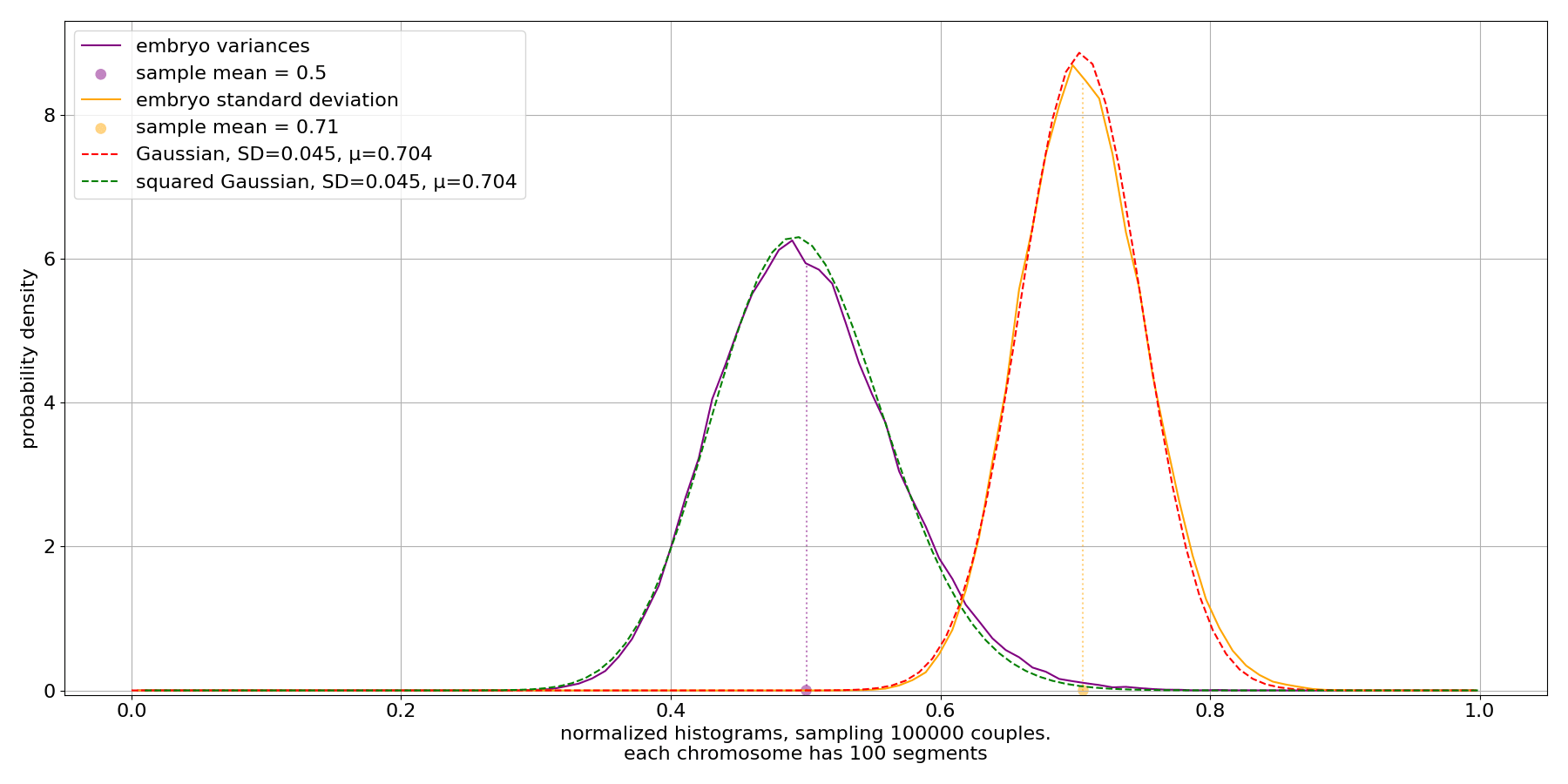
The "squared gaussian" is the distribution of the square of the SD value. The formula used is
where
by eyeballing it. Here is a gaussian distribution. I think this is a noncentral chi-squared distribution.
What is the maximum scoring embryo from one couple?
We have some handle on the distribution, across couples, of the distribution of a couple's embryos. The embryo-distribution is roughly a gaussian. (We can basically ignore the mean because, as we saw, the embryo distribution doesn't appreciably change with the couple's genome mean. This relies on PGSes having many hundreds of target alleles, which is already true as of 2024.) The embryo-distribution gaussian has SD sampled from roughly a gaussian with mean , SD .
So, to model a couple, we first sample from the embryo-distribution-SD gaussian, to get an SD σ. Then, to sample embryos, we sample from a gaussian with mean 0 and SD σ. This represents not the raw score of the embryo, but rather the difference between the embryo and the couple's mean score. To further standardize, we can instead model sampling from a standard normal distribution (mean = 0, SD = 1). This represents "couple SDs", i.e. SDs of this specific couple's embryos. Then, whatever the maximum is, we can convert that back into real SDs (i.e., SDs of the general population) by multiplying by σ, and then adding the couple's mean.
Now we just need to know the maximum sample from a standard normal. For this, see the Simple Selection section of "The power of selection".
The basic answers we want can be read off the last two graphs in the "Maximum sample out of " section. Those graphs show the value of the maximum sample from a standard normal, with the number of samples maxxed over shown on the x-axis and the SD value shown on the y-axis. Here's the logscale one:
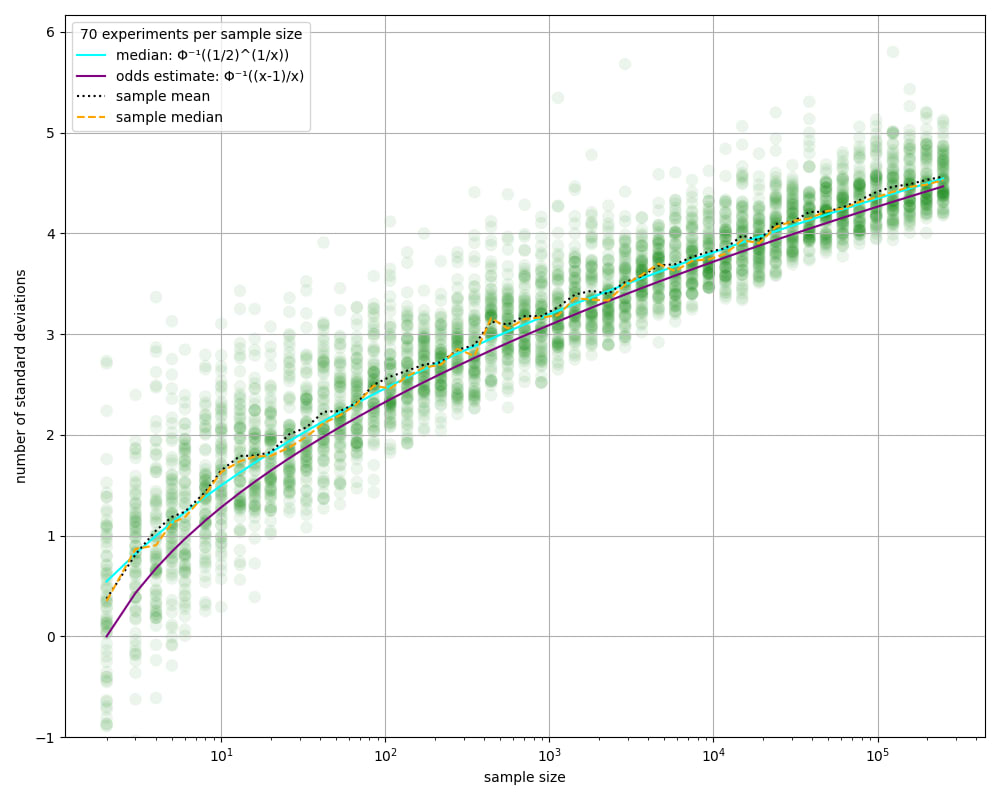
To convert to the values for one couple's embryos, we have to adjust to the couple's embryo-distribution. In the modal case, the couple's embryos are distributed like a gaussian with SD about .7. So we have to multiply the values we read on those graphs by .7. E.g. sampling about 30 embryos would give around SDs of selection power. This says how much higher the embryo scores, compared to the mean of the parents's scores.
Looking at the logscale table, and considering a very optimistic case where IVO is available, we see that sampling 100,000 embryos would give roughly SDs of selection power. However, this is only raw selection power. The resulting embryo is 3 SDs extreme, as measured by whatever we were selecting for. But our PGS is only partially correlated with the trait/s we're selecting for. As a toy example, suppose we only select for IQ, and we have a PGS that correlates .5 with the trait (currently there are IQ PGSes that correlate around .3 or .4). So we multiply the raw selection power of 3 SDs by .5 to get 1.5 SDs of effective selection power. Thus, even with very many embryos (requiring IVO), simple embryo selection only gets twenty-something IQ points. (And in reality you would use some selection power to select for other traits, e.g. to decrease disease risk.)
These estimates are reasonable rule-of-thumb estimates. But it would also be good to understand a bit more about the distribution of maximum scores. Reason 1: To get a sense of how much variation to expect from a real-life implementation, e.g. to not be too disappointed with an outcome a little below the mean outcome. Reason 2: Some of this understanding will transfer to understanding the results of other sampling procedures, e.g. sampling haploids.
Here's a graph showing the results of taking a couple, getting some embryos from them, and then taking the highest-scoring one. The graph shows the results from taking 4 embryos, or taking 30 embryos; and shows the results for a set of random couples; and for a set of couples selected to have embryo scores distributed scores with standard deviation more than two embryo-SD-SDs greater than the mean embryo-SD (); and for a set with embryo-SD less than two embryo-SD-SDs under the mean embryo-SD (). (We're always looking at the difference between the embryo score and the couple's mean score; or in other words, we're conditioning on all the couples having mean score 0 SDs. These distributions are basically the same for any couple, since embryo variance isn't much affected by the couple's scores for highly polygenic traits.)
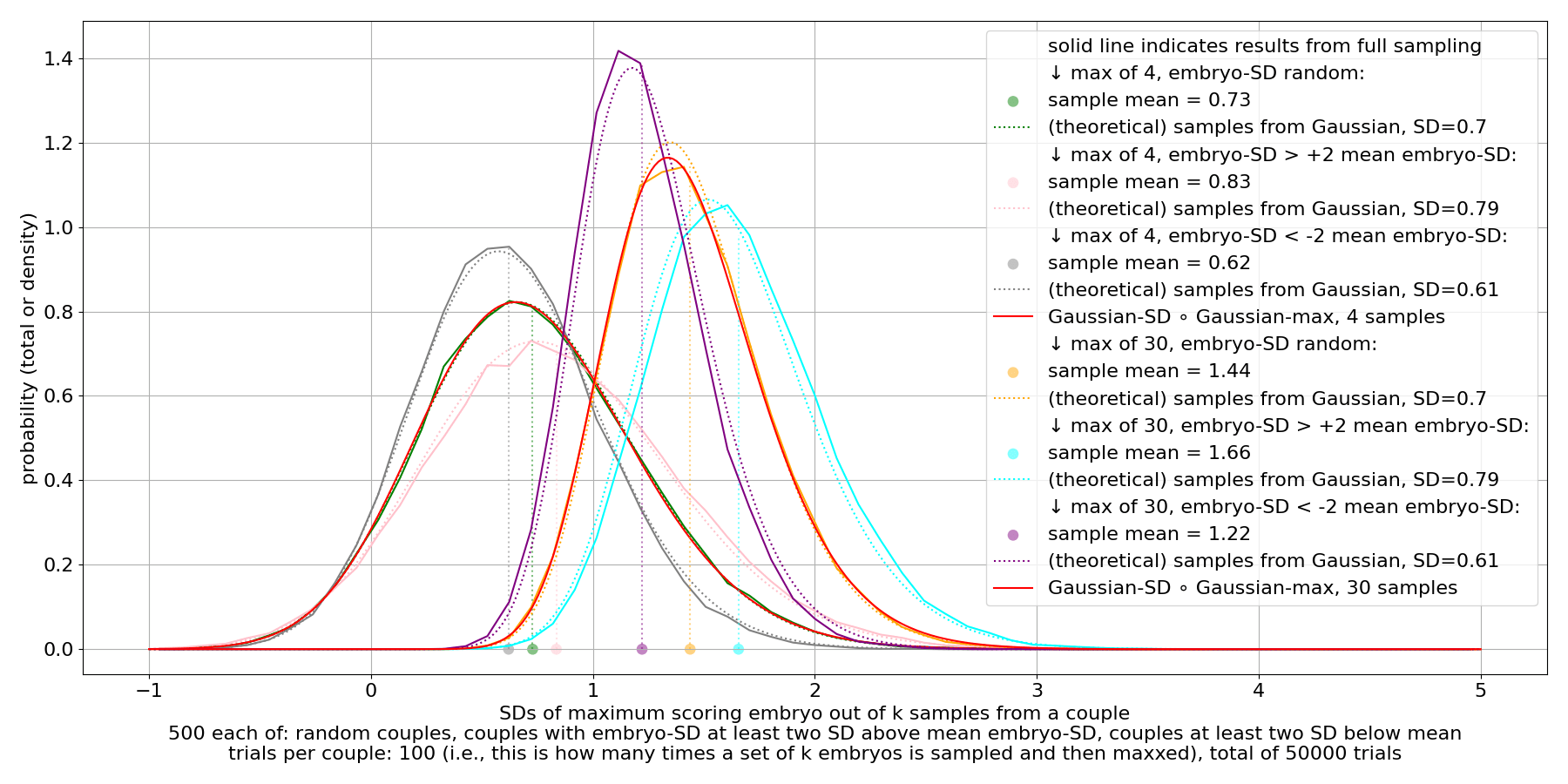
We see that there's substantial variance in the max. This persists even for very large sample counts.
It's computationally expensive to do this sampling for large sampling counts. But we can use the theoretical predictions, described in the "Maximum sample out of " section. The simplest way is to use the max-gaussian distribution, for a gaussian with SD equal to the mean embryo-SD, . This works reasonably well; these are the dotted lines in the above graph. For larger sample counts, this starts to diverge a little from the sampled results. We can see this by using another cheap approximation: Instead of actually sampling embryos, we can use the approximation of the embryo distribution as a gaussian. So we sample couples and compute their embryo SD, and then to simulate sampling embryos, we sample from a gaussian with that SD and with mean 0. Here's a graph showing this sampling, and showing the simple max-gaussian distributions:
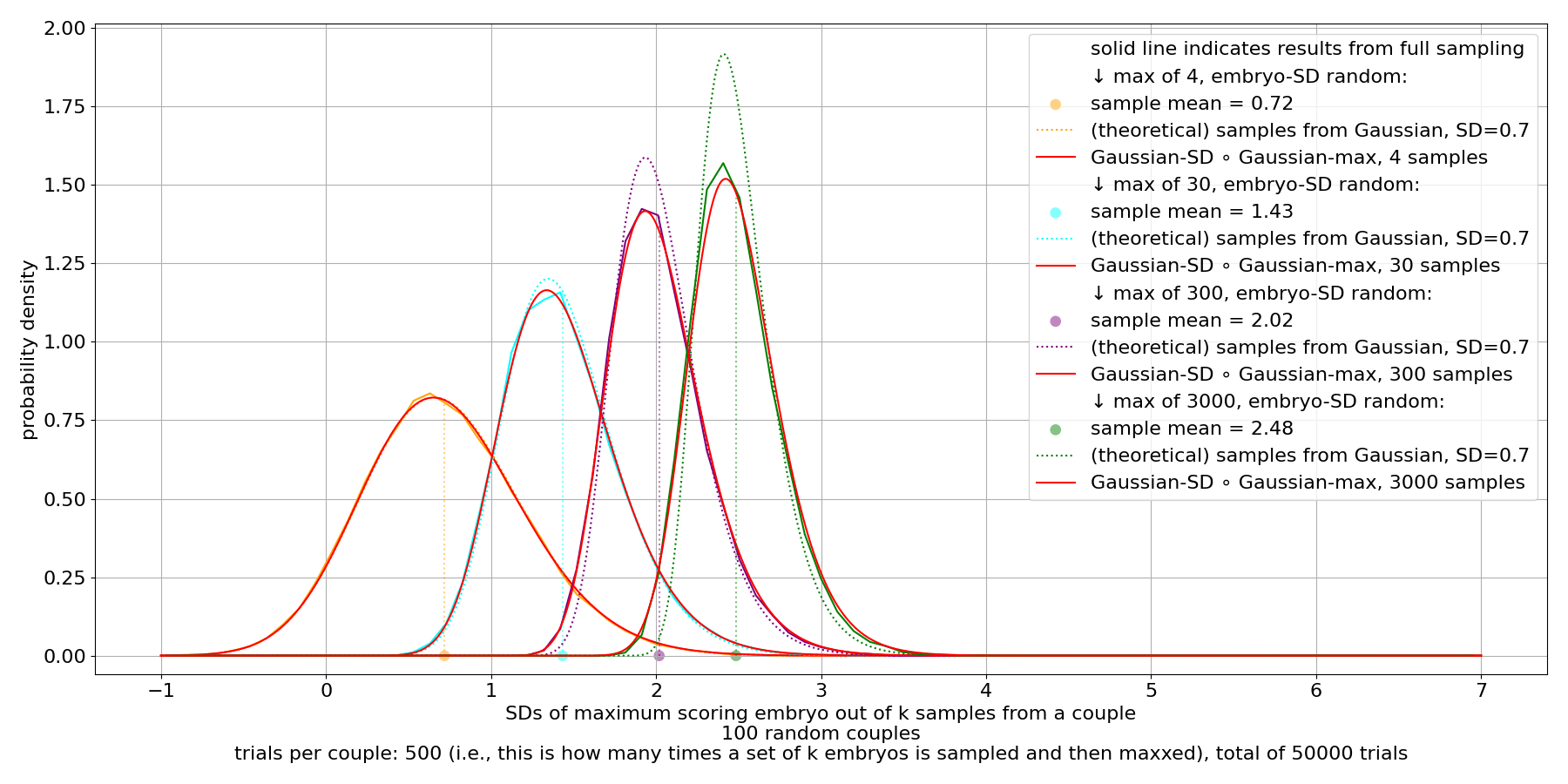
We see that for larger sample counts, the simple max-gaussian is significantly tighter of a distribution compared to the actual samples. That's because the actual samples are the max from gaussians, but the gaussians have SD that has variance. We can better approximate the distribution by modeling that process. Instead of using one max-gaussian with mean SD, we take a mixture of max-gaussians. We weight the max-gaussian for according to the probability of in the gaussian that describes embryo-SDs, i.e. mean , SD . The red lines in the above graphs show a rough approximation of this distribution (a mixture of 100 max-gaussians). We see that it's a very close fit, practically, and it's much faster to compute than sampling.
Using this mixture, we can see the results of maxxing over a set of embryos from one couple, for very large numbers of embryos:
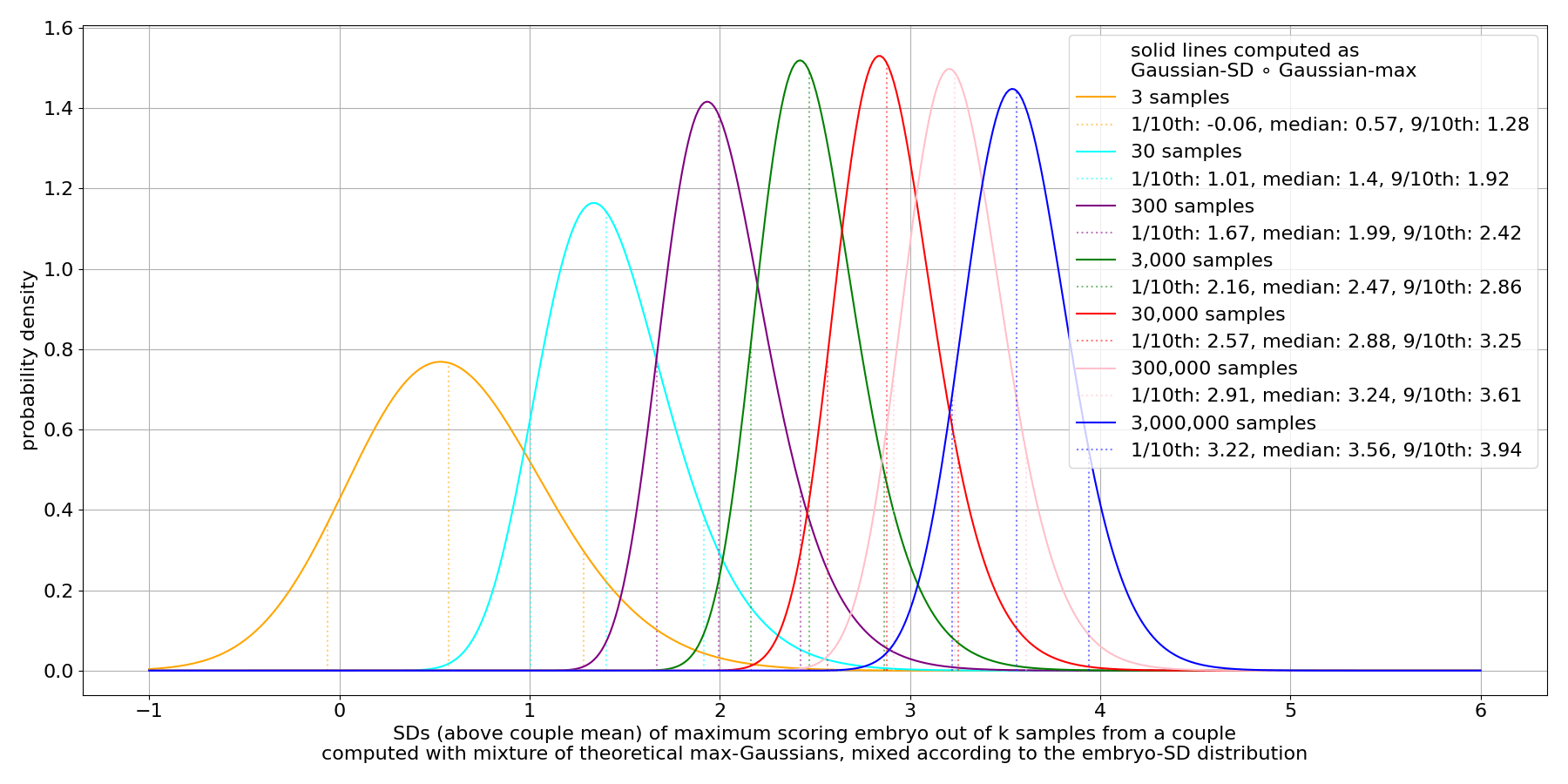
This plot shows the lower decile, the median, and the upper decile, as dotted lines; and shows the mixed-max-gaussian distribution for various . (Notice that the upper decile, the rightmost of the three dotted lines in each color, for the max- distribution tends to be close to the median line (the middle of the dotted lines) for the max- distribution. This isn't a coincidence: The upper decile is about 1 in 10 rare, so getting an upper decile outcome happens roughly when you sample ten times. Sampling 10 times from the max- distribution is equivalent to sampling once from the max- distribution.)
Plotting with deciles shaded, to get a sense for the dispersion:
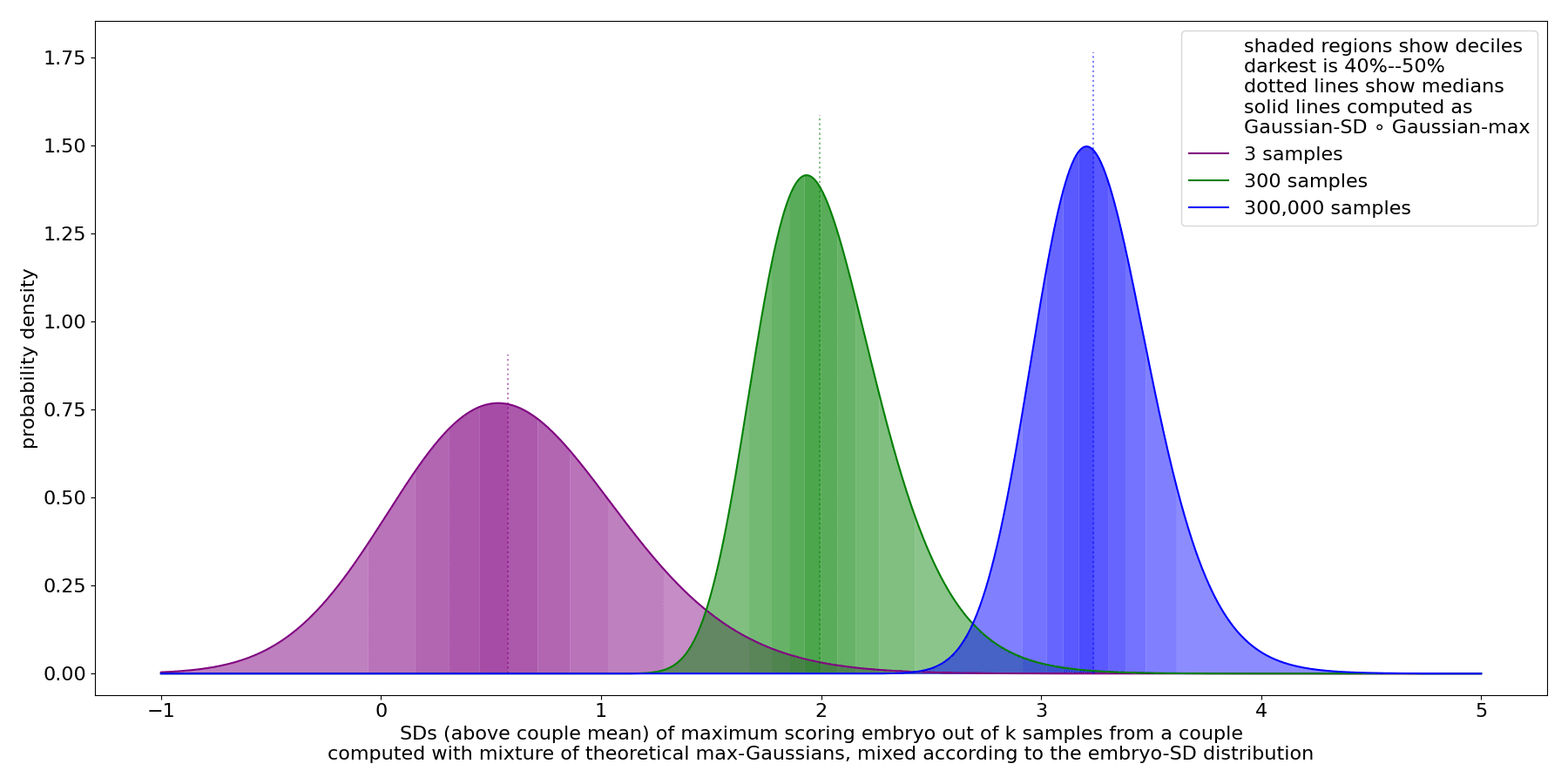
Putting together our numbers in a table:
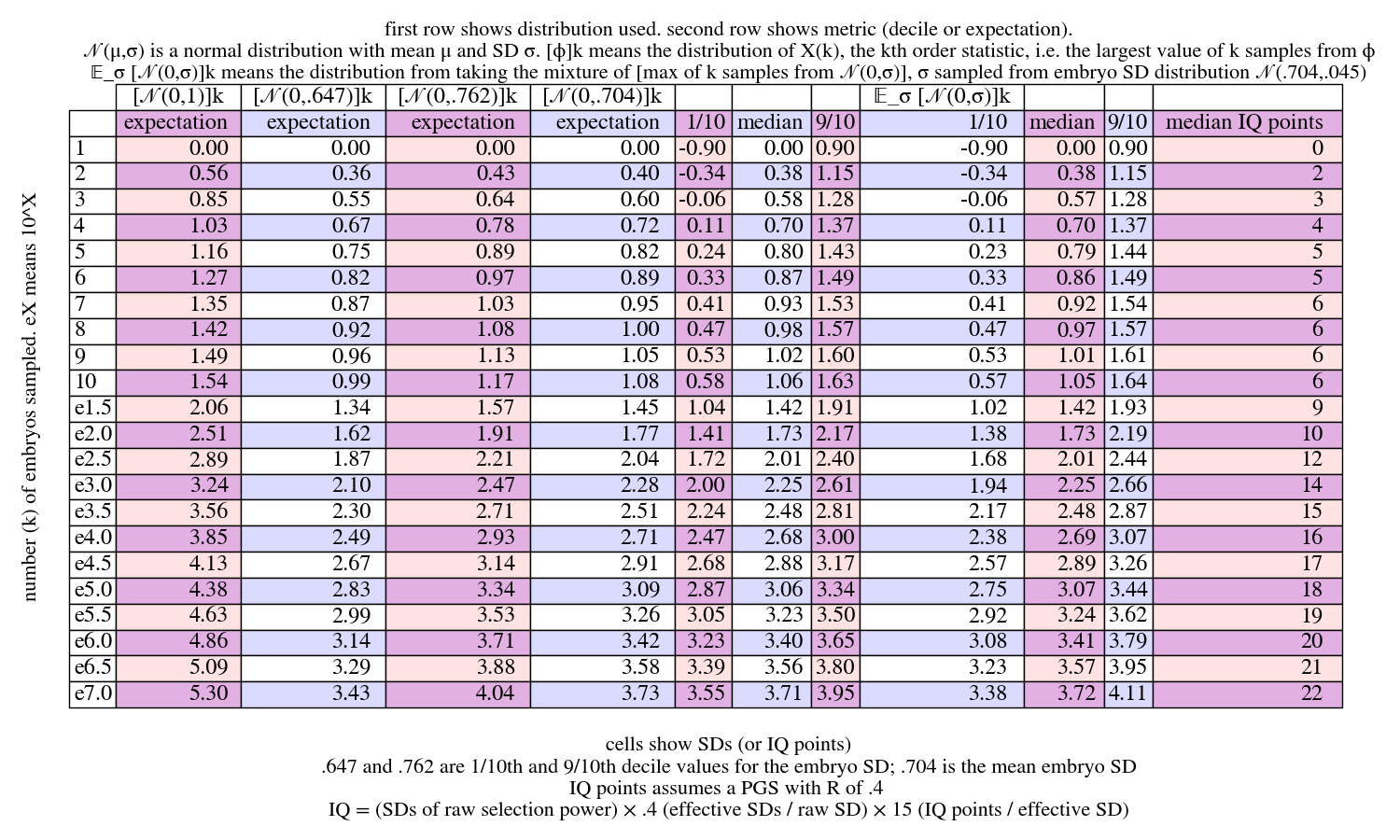
This shows the SDs above the couple mean of the highest-scoring embryo of embryos sampled from a couple, with various approximations. The first few columns just show normal distributions with SD being , or the mean embryo SD , or the bottom or top decile embryo SDs. These distributions represent the distribution for a couple with the corresponding embryo SD, after sequencing the parents and calculating that embryo SD. (The various are just scaled versions of , so their statistics are also just scaled versions. E.g. a number in the expectation column for is just times the corresponding number in the column. This is not true for the mixture distribution.)
The distribution labeled is
i.e. the mixture of -max-embryo distributions, where is sampled from the embryo-SD distribution . This represents the distribution for a couple's ultimate outcome from maxxing over embryos, before we know the parents's genomes—i.e., we sample a couple from the population and then also sample embryos and take the highest scoring.
Takeaways:
- For most purposes, we can just use the mixture distribution . The median of the mixture distribution is similar to the mean and to the central tendency of the narrower distribution that uses the mean embryo-SD. The mixture is wider.
- For convenience we can just use the median as our estimate. It's easier to compute, it's a slightly conservative estimate, and it's more comparable to the deciles. But the -max distribution is skewed high, so the expectation is a little higher than the median.
- We quickly hit a big wall when sampling more and more. Going from 1 to 100 embryos gives +10 IQ points; going from 100 to 1,000,000 also gives +10 IQ points.
- The IQ estimates, or similar estimates for any other trait, depend on the PGS for that trait. If the PGS has an R value of .6 instead of .4, you get +50% the effect.
To get a better sense of the range of outcomes, we can zoom in on this mixture distribution:
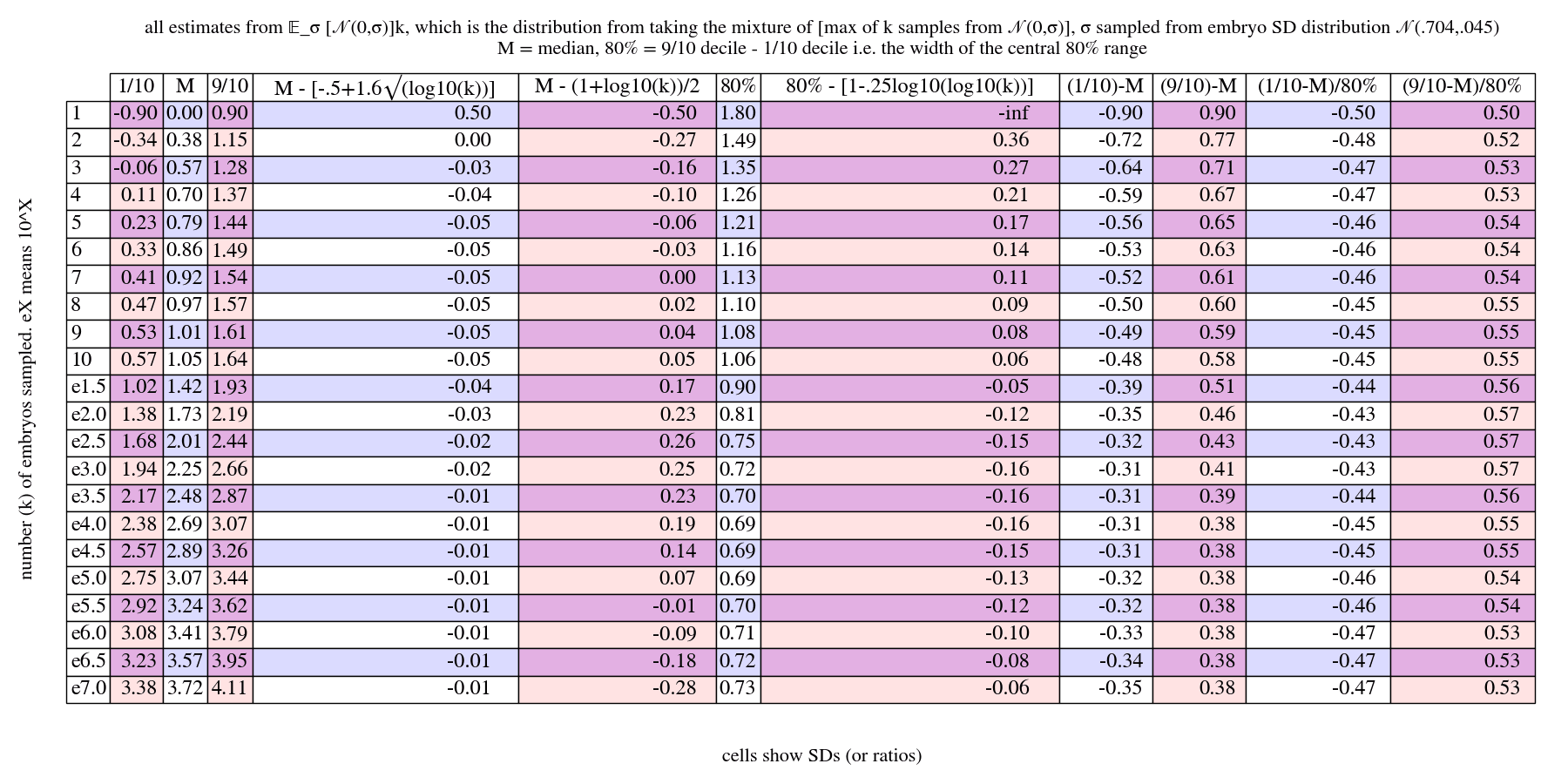
Takeaways:
- The median is very roughly , to within about . Much more closely, it's roughly . So for example with , i.e. 100 embryos, we get , close to the real value of .
- The 80% range between the bottom 1/10th and the top 1/10th is very very roughly 1. A closer approximation is . So for we get , sorta close to the real value of . But probably best to just remember that the range is greater than 1 SD for very small , 1 SD for smallish , and about for .
- General note: , and .
- The split of the 80% interval between up to median, vs. median up to , starts at 50/50 and gets more lopsided as grows, going to almost 40/60 for around 100, and then returning to an evenish split. So we can basically think of it as 50/50, or 45/55 to be a bit more precise. We'll write e.g. that for , the result is about for the 80% range, even though really it's sometimes more like a 45/55 split. A simple qualitative summary to remember is that the results of embryo selection skew so that there are a bit more very-high outcomes than very-low outcomes. (I think what's happening is that, at first, the spread of the gaussian-max dominates. That spread decreases with more samples. But then the embryo-SD mixture spread starts dominating, and that is symmetric and doesn't decrease.)
Converting to IQ points, and showing the half-widths of 80% intervals:
Takeaways:
- The wall is real. With 10 embryos you get 6 IQ points. Up to 1000, each order of magnitude gets you +4 IQ points. Thereafter, each order of magnitude gets you +2 IQ points. So again: first OOM is 6 points, next two OOMs are 4 points each, further OOMs are 2 points each.
- Different couples will have outcomes that vary by about 3 IQ points up or down. (Again, all relative to the couple's mean.)
Appendix: Variation in chromosome length
(For this section, you can follow along in a python interpreter by importing this code, or just the lines at the beginning that define some lengths: https://gist.github.com/tsvibt/89cec1b4fd7d54be04ba34a0059bcc4c.)
The issue with chromosome lengths
Human chromosomes vary a lot in length:
[(Figure 1.3 from Gallegos (2022)[109:1].)]
This corresponds to a large variation in the number of base pairs in the chromosomes. The largest chromosome, chromosome 1, is over five times longer than the shortest, chromosome 21:
>>> max(chromosome_lengths.values())
247249719
>>> min(chromosome_lengths.values())
46944323
>>> max(chromosome_lengths.values()) / min(chromosome_lengths.values())
5.266871544829819
That means chromosome 1 has five times as much variance as chromosome 21. In several contexts in this article, we ignore this, and just pretend that all the chromosomes have the same length. For example, for simple chromosome selection of a haploid genome, we compute:
The is a very rough estimate of the power, in SDs, of selecting the best 1 of 2 samples from a standard gaussian. We get to select 22 chromosomes (ignoring the sex chromosome). And then we pretend that each chromosome is the length of the whole genome, so that it has of the variance. That would mean a chromosome-SD is of a whole-genome-SD.
Certainly in the limit, if one chromosome were really huge, like 99% of the genome, and all the others were tiny, we'd get very little selection power. The tiny chromosomes wouldn't matter, and the big chromosome would only give us about .5 or .6 SDs of selection power (divided by ); it'd basically be gamete selection, were you only pick from 2 gametes.
Do chromosome lengths matter much?
What about for the actual chromosome lengths? Our rough formula, generalized, is this:
The summation can be rewritten as:
Or:
We compute:
>>> sum(np.sqrt(x) for x in [1/46]*23)
3.391164991562635
>>> sum(np.sqrt(x) for x in diploid_chromosome_fractions.values())
3.309001050655313
The first command gives the chromosome selection factor for 23 chromosomes of equal length. The second command gives the factor for 23 chromosomes of realistic length (where the 23rd chromosome has length the average of the lengths of the X and Y chromosomes).
The difference is less than 3%!
Here's a graph showing various ways of estimating chromosome selection, from "The power of selection":
Yes, you can see the difference between the estimates based on chromosome length and the ones that ignore it. You can also see how small the difference is.
Why don't chromosome lengths matter much?
The variance scales linearly with length, but the SDs scale as the square root of length. The longest chromosome is over five times as long as the shortest, but its SD scale is only a little more than two times the scale of the shortest:
>>> np.sqrt(max(chromosome_lengths.values()) / diploid_total_length )
0.20388105167623205
>>> np.sqrt(min(chromosome_lengths.values()) / diploid_total_length )
0.08883835371940446
To say it another way, the marginal returns on more SDs diminish with the square root, as you increase the length of the chromosome. Or to say it another another way, it's more important to select on the chromosome, than that it be long.
As a touchpoint: compare selecting just two chromosomes. They could be the same lengths, , or one could be 5 times longer, . For the former, the chromosome selection factor is . For the latter, the factor is . Not even a 10% difference.
In fact, what about going all the way to ? Then the factor is 1, which is about 70% of 1.41. In other words, completely merging two chromosomes of equal length is a 30% hit to their combined selection factor.
When does it make a big difference? Suppose we start with two chromosomes, each half the size of the whole genome so to speak, and then shift length from one to the other. Now we've removed some fraction of one and added it to the other, so we have a and a . As we move all the mass to the longer one, what's the selection factor?
We can plot this by giving the expression
plot sqrt(z - z f) + sqrt(z + z f) for z=.5, f from 0 to 1
to WolframAlpha. We get:
(The graph looks the same, but scaled, if you use .)
Another perspective: Say you're trying to decrease the power of selection by shoveling chromosomes from shorter chromosomes to longer ones, increase the length inequality. At first, starting from equal lengths, you basically aren't doing anything: you're moving in opposite directions starting from the same point on a smooth curve, so at first the derivatives are just each other's negations. As the inequality increases, the difference in derivatives increases. But because of the shape of the square root, the losses from shortening the short guy only get much worse than the gains from lengthening the long guy when the short guy is pretty comparatively short.
To formalize this a bit, we can look at the derivative . Again suppose we start with both chromosomes of length , and then shift length from one to the other. If we keep doing that, what's the derivative? We plot this by giving the expression
plot (1/(2 sqrt(z - z f)) - 1/(2 sqrt(z + z f))) for z=.5, f from 0 to 1
to WolframAlpha. We get:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The rate of loss of SDs, as we shift length, starts at zero and climbs. But it only really spikes well toward the end, when we've almost eliminated the short chromosome.
Ignoring the 23rd chromosome
We did the above computations with 23 chromosomes. What if we ignore the 23rd chromosome? We get:
>>> sum(np.sqrt(x) for i,x in enumerate([1/46]*23) if i<22)
3.2437230354077378
>>> sum(np.sqrt(x) for i,x in enumerate(diploid_chromosome_fractions.values()) if i<22)
3.175290973758385
So with 23 chromosomes, the equal-length estimate is 3.39 vs. varying-length says 3.31. With 22 chromosomes, the equal-length estimate is 3.24 vs. varying-length says 3.17. I'd just as happily ignore the 23rd chromosome simply out of convenience to avoid complications (e.g., recombination works differently for the 23rd chromosome). Throwing out the 23rd chromosome from being selected over has more effect than taking lengths into account, and it's still less than 5%!
The upshot is that, for most selection purposes, it scarcely matters that the chromosomes are different lengths. Certainly if translated to real-world applications, none of our estimates are accurate to within 5% anyway.
Acknowledgements
Thanks to Sam Eisenstat for discussions of some of the mathematical ideas in this article. Thanks to kman [LW · GW] for discussion of GV methods.
License
All content (such as diagrams) that isn't from an external source is shareable under the CC BY-SA 4.0 license (https://creativecommons.org/licenses/by-sa/4.0/) with attribution to the Berkeley Genomics Project, 2025.
Gwern Branwen. ‘Embryo Selection For Intelligence’, 22 January 2016. https://gwern.net/embryo-selection. ↩︎ ↩︎
GeneSmith, and kman. ‘How to Make Superbabies’, 19 February 2025. https://www.lesswrong.com/posts/DfrSZaf3JC8vJdbZL/how-to-make-superbabies [LW · GW]. ↩︎ ↩︎
Miller, Joshua D., and Donald R. Lynam. ‘Understanding Psychopathy Using the Basic Elements of Personality’. Social and Personality Psychology Compass 9, no. 5 (2015): 223–37. https://doi.org/10.1111/spc3.12170. ↩︎
Metacelsus. ‘Epigenetics of the Mammalian Germline’. Substack newsletter. De Novo (blog), 23 December 2022. https://denovo.substack.com/p/epigenetics-of-the-mammalian-germline. ↩︎ ↩︎
Kelsey, Gavin, and Robert Feil. ‘New Insights into Establishment and Maintenance of DNA Methylation Imprints in Mammals’. Philosophical Transactions of the Royal Society B: Biological Sciences 368, no. 1609 (5 January 2013): 20110336. https://doi.org/10.1098/rstb.2011.0336. ↩︎ ↩︎ ↩︎ ↩︎
Jima, Dereje D., David A. Skaar, Antonio Planchart, Alison Motsinger-Reif, Sebnem E. Cevik, Sarah S. Park, Michael Cowley, et al. ‘Genomic Map of Candidate Human Imprint Control Regions: The Imprintome’. Epigenetics 17, no. 13 (9 December 2022): 1920–43. https://doi.org/10.1080/15592294.2022.2091815. ↩︎
Ishihara, Teruhito, Oliver W. Griffith, Shunsuke Suzuki, and Marilyn B. Renfree. ‘Presence of H3K4me3 on Paternally Expressed Genes of the Paternal Genome From Sperm to Implantation’. Frontiers in Cell and Developmental Biology 10 (10 March 2022). https://doi.org/10.3389/fcell.2022.838684. ↩︎
Tanaka, Atsushi, and Seiji Watanabe. ‘How to Improve the Clinical Outcome of Round Spermatid Injection (ROSI) into the Oocyte: Correction of Epigenetic Abnormalities’. Reproductive Medicine and Biology 22, no. 1 (9 February 2023): e12503. https://doi.org/10.1002/rmb2.12503. ↩︎ ↩︎ ↩︎ ↩︎
Liu, Zhen, Yijun Cai, Yan Wang, Yanhong Nie, Chenchen Zhang, Yuting Xu, Xiaotong Zhang, et al. ‘Cloning of Macaque Monkeys by Somatic Cell Nuclear Transfer’. Cell 172, no. 4 (8 February 2018): 881-887.e7. https://doi.org/10.1016/j.cell.2018.01.020. ↩︎ ↩︎ ↩︎
Schnleke, A E, K H S Campbell, Ian Wilmut, J McWhir, and AJ Kind. ‘Viable Offspring Derived from Fetal and Adult Mammalian Cells’, 1997, https://www.nature.com/articles/385810a0.pdf. ↩︎
Olsson, P. Olof, Yeon Woo Jeong, Yeonik Jeong, Mina Kang, Gang Bae Park, Eunji Choi, Sun Kim, Mohammed Shamim Hossein, Young-Bum Son, and Woo Suk Hwang. ‘Insights from One Thousand Cloned Dogs’. Scientific Reports 12, no. 1 (1 July 2022): 11209. https://doi.org/10.1038/s41598-022-15097-7. ↩︎
Smith, Lc, J Suzuki Jr, Ak Goff, F Filion, J Therrien, Bd Murphy, Hr Kohan-Ghadr, et al. ‘Developmental and Epigenetic Anomalies in Cloned Cattle’. Reproduction in Domestic Animals 47, no. s4 (2012): 107–14. https://doi.org/10.1111/j.1439-0531.2012.02063.x. ↩︎ ↩︎
Liehr, Thomas. ‘Cases with Uniparental Disomy’, 2025. https://cs-tl.de/DB/CA/UPD/0-Start.html. ↩︎
Morpheus. ‘What Uniparental Disomy Tells Us About Improper Imprinting in Humans’, 28 March 2025. https://www.lesswrong.com/posts/49Y2WDxStNBWMCBku/what-uniparental-disomy-tells-us-about-improper-imprinting [LW · GW]. ↩︎
Nayernia, Karim, Jessica Nolte, Hans W. Michelmann, Jae Ho Lee, Kristina Rathsack, Nadja Drusenheimer, Arvind Dev, et al. ‘In Vitro-Differentiated Embryonic Stem Cells Give Rise to Male Gametes That Can Generate Offspring Mice’. Developmental Cell 11, no. 1 (1 July 2006): 125–32. https://doi.org/10.1016/j.devcel.2006.05.010. ↩︎
Li, Zhi-kun, Li-bin Wang, Le-yun Wang, Xue-han Sun, Ze-hui Ren, Si-nan Ma, Yu-long Zhao, et al. ‘Adult Bi-Paternal Offspring Generated through Direct Modification of Imprinted Genes in Mammals’. Cell Stem Cell, 28 January 2025. https://doi.org/10.1016/j.stem.2025.01.005. ↩︎ ↩︎
Li, Zhikun, Haifeng Wan, Guihai Feng, Leyun Wang, Zhengquan He, Yukai Wang, Xiu-Jie Wang, Wei Li, Qi Zhou, and Baoyang Hu. ‘Birth of Fertile Bimaternal Offspring Following Intracytoplasmic Injection of Parthenogenetic Haploid Embryonic Stem Cells’. Cell Research 26, no. 1 (January 2016): 135–38. https://doi.org/10.1038/cr.2015.151. ↩︎
Mitalipov, Shoukhrat M., Richard R. Yeoman, Kevin D. Nusser, and Don P. Wolf. ‘Rhesus Monkey Embryos Produced by Nuclear Transfer from Embryonic Blastomeres or Somatic Cells1’. Biology of Reproduction 66, no. 5 (1 May 2002): 1367–73. https://doi.org/10.1095/biolreprod66.5.1367. ↩︎ ↩︎
Luciano, Alberto Maria, and Valentina Lodde. ‘Changes of Large-Scale Chromatin Configuration During Mammalian Oocyte Differentiation’. In Oogenesis, edited by Giovanni Coticchio, David F. Albertini, and Lucia De Santis, 93–108. London: Springer, 2013. https://doi.org/10.1007/978-0-85729-826-3_7. ↩︎
Trasler, Jacquetta M. ‘Gamete Imprinting: Setting Epigenetic Patterns for the next Generation’. Reproduction, Fertility and Development 18, no. 2 (2006): 63. https://www.publish.csiro.au/rd/Fulltext/RD05118. ↩︎ ↩︎ ↩︎ ↩︎
Adam Green. ‘A Future History of Biomedical Progress’, 1 August 2022. https://markovbio.github.io/biomedical-progress/. ↩︎
Baysoy, Alev, Zhiliang Bai, Rahul Satija, and Rong Fan. ‘The Technological Landscape and Applications of Single-Cell Multi-Omics’. Nature Reviews Molecular Cell Biology 24, no. 10 (October 2023): 695–713. https://doi.org/10.1038/s41580-023-00615-w. ↩︎ ↩︎
Guo, Jingtao, Edward J. Grow, Hana Mlcochova, Geoffrey J. Maher, Cecilia Lindskog, Xichen Nie, Yixuan Guo, et al. ‘The Adult Human Testis Transcriptional Cell Atlas’. Cell Research 28, no. 12 (December 2018): 1141–57. https://doi.org/10.1038/s41422-018-0099-2. ↩︎
Sohni, Abhishek, Kun Tan, Hye-Won Song, Dana Burow, Dirk G. de Rooij, Louise Laurent, Tung-Chin Hsieh, et al. ‘The Neonatal and Adult Human Testis Defined at the Single-Cell Level’. Cell Reports 26, no. 6 (5 February 2019): 1501-1517.e4. https://doi.org/10.1016/j.celrep.2019.01.045. ↩︎
Wang, Si, Yuxuan Zheng, Jingyi Li, Yang Yu, Weiqi Zhang, Moshi Song, Zunpeng Liu, et al. ‘Single-Cell Transcriptomic Atlas of Primate Ovarian Aging’. Cell 180, no. 3 (6 February 2020): 585-600.e19. https://doi.org/10.1016/j.cell.2020.01.009. ↩︎
Jones, Andrea S. K., D. Ford Hannum, Jordan H. Machlin, Ansen Tan, Qianyi Ma, Nicole D. Ulrich, Yu-chi Shen, et al. ‘Cellular Atlas of the Human Ovary Using Morphologically Guided Spatial Transcriptomics and Single-Cell Sequencing’. Science Advances 10, no. 14 (5 April 2024): eadm7506. https://doi.org/10.1126/sciadv.adm7506. ↩︎
Saitou, Mitinori, and Hidetaka Miyauchi. ‘Gametogenesis from Pluripotent Stem Cells’. Cell Stem Cell 18, no. 6 (June 2016): 721–35. https://doi.org/10.1016/j.stem.2016.05.001. ↩︎ ↩︎ ↩︎ ↩︎
Saitou, Mitinori, and Katsuhiko Hayashi. ‘Mammalian in Vitro Gametogenesis’. Science 374, no. 6563 (October 2021): eaaz6830. https://doi.org/10.1126/science.aaz6830. ↩︎ ↩︎ ↩︎ ↩︎ ↩︎
Robinson, Meghan, Sydney Sparanese, Luke Witherspoon, and Ryan Flannigan. ‘Human in Vitro Spermatogenesis as a Regenerative Therapy — Where Do We Stand?’ Nature Reviews Urology 20, no. 8 (August 2023): 461–79. https://doi.org/10.1038/s41585-023-00723-4. ↩︎ ↩︎ ↩︎
Gilchrist, Daniel A. ‘Meiosis’. Accessed 2 March 2025. https://www.genome.gov/genetics-glossary/Meiosis. ↩︎
Campbell, Alison, and Walid Maalouf. Mastering Clinical Embryology: Good Practice, Clinical Biology, Assisted Reproductive Technologies, and Advanced Laboratory Skills. CRC Press, 2024, https://www.google.com/books/edition/Mastering_Clinical_Embryology/xkb3EAAAQBAJ. ↩︎ ↩︎
Sharma, Rakesh, and Ashok Agarwal. ‘Spermatogenesis: An Overview’. In Sperm Chromatin: Biological and Clinical Applications in Male Infertility and Assisted Reproduction, edited by Armand Zini and Ashok Agarwal, 19–44. New York, NY: Springer, 2011. https://doi.org/10.1007/978-1-4419-6857-9_2. ↩︎
Mtango, Namdori R., Santhi Potireddy, and Keith E. Latham. ‘Chapter 7 Oocyte Quality and Maternal Control of Development’. In International Review of Cell and Molecular Biology, 268:223–90. Academic Press, 2008. https://doi.org/10.1016/S1937-6448(08)00807-1. ↩︎
Pors, S. E., D. Nikiforov, J. Cadenas, Z. Ghezelayagh, Y. Wakimoto, L. A. Z. Jara, J. Cheng, et al. ‘Oocyte Diameter Predicts the Maturation Rate of Human Immature Oocytes Collected Ex Vivo’. Journal of Assisted Reproduction and Genetics 39, no. 10 (October 2022): 2209–14. https://doi.org/10.1007/s10815-022-02602-0. ↩︎
Champroux, Alexandre, Julie Cocquet, Joëlle Henry-Berger, Joël R. Drevet, and Ayhan Kocer. ‘A Decade of Exploring the Mammalian Sperm Epigenome: Paternal Epigenetic and Transgenerational Inheritance’. Frontiers in Cell and Developmental Biology 6 (15 May 2018). https://doi.org/10.3389/fcell.2018.00050. ↩︎ ↩︎
Yoon, Sook-Young, and Rafael A. Fissore. ‘Release of Phospholipase C Ζand [Ca2+]i Oscillation-Inducing Activity during Mammalian Fertilization’, 1 November 2007. https://doi.org/10.1530/REP-07-0259. ↩︎
Saitou, Mitinori, and Katsuhiko Hayashi. ‘Mammalian in Vitro Gametogenesis’. Science 374, no. 6563 (October 2021): eaaz6830. https://doi.org/10.1126/science.aaz6830. ↩︎ ↩︎
Tang, Walfred W. C., Toshihiro Kobayashi, Naoko Irie, Sabine Dietmann, and M. Azim Surani. ‘Specification and Epigenetic Programming of the Human Germ Line’. Nature Reviews Genetics 17, no. 10 (October 2016): 585–600. https://doi.org/10.1038/nrg.2016.88. ↩︎ ↩︎
Gkountela, Sofia, Ziwei Li, John J. Vincent, Kelvin X. Zhang, Angela Chen, Matteo Pellegrini, and Amander T. Clark. ‘The Ontogeny of cKIT+ Human Primordial Germ Cells Proves to Be a Resource for Human Germ Line Reprogramming, Imprint Erasure and in Vitro Differentiation’. Nature Cell Biology 15, no. 1 (January 2013): 113–22. https://doi.org/10.1038/ncb2638. ↩︎
Takahashi, Kazutoshi, and Shinya Yamanaka. ‘Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors’. Cell 126, no. 4 (25 August 2006): 663–76. https://doi.org/10.1016/j.cell.2006.07.024. ↩︎ ↩︎ ↩︎
Irie, Naoko, Leehee Weinberger, Walfred W.C. Tang, Toshihiro Kobayashi, Sergey Viukov, Yair S. Manor, Sabine Dietmann, Jacob H. Hanna, and M. Azim Surani. ‘SOX17 Is a Critical Specifier of Human Primordial Germ Cell Fate’. Cell 160, no. 1–2 (15 January 2015): 253–68. https://doi.org/10.1016/j.cell.2014.12.013. ↩︎ ↩︎ ↩︎
Sasaki, Kotaro, Shihori Yokobayashi, Tomonori Nakamura, Ikuhiro Okamoto, Yukihiro Yabuta, Kazuki Kurimoto, Hiroshi Ohta, et al. ‘Robust In Vitro Induction of Human Germ Cell Fate from Pluripotent Stem Cells’. Cell Stem Cell 17, no. 2 (6 August 2015): 178–94. https://doi.org/10.1016/j.stem.2015.06.014. ↩︎ ↩︎ ↩︎
Panula, Sarita, Jose V. Medrano, Kehkooi Kee, Rosita Bergström, Ha Nam Nguyen, Blake Byers, Kitchener D. Wilson, et al. ‘Human Germ Cell Differentiation from Fetal- and Adult-Derived Induced Pluripotent Stem Cells’. Human Molecular Genetics 20, no. 4 (15 February 2011): 752–62. https://doi.org/10.1093/hmg/ddq520. ↩︎
Yamashiro, Chika, Kotaro Sasaki, Shihori Yokobayashi, Yoji Kojima, and Mitinori Saitou. ‘Generation of Human Oogonia from Induced Pluripotent Stem Cells in Culture’. Nature Protocols 15, no. 4 (April 2020): 1560–83. https://doi.org/10.1038/s41596-020-0297-5. ↩︎ ↩︎ ↩︎
Trasler, Jacquetta M. ‘Epigenetics in Spermatogenesis’. Molecular and Cellular Endocrinology, Proceedings of the 15th Workshop on Molecular and Cellular Endocrinology of the Testis, 306, no. 1 (10 July 2009): 33–36. https://doi.org/10.1016/j.mce.2008.12.018. ↩︎ ↩︎
Keser, Vanessa. ‘Lokalizacija proteina kontrolne točke diobenog vretena u mišjim spermatocitama’. Info:eu-repo/semantics/bachelorThesis, University of Zagreb. Faculty of Food Technology and Biotechnology. Department of Biochemical Engineering. Laboratory for Biology and Microbial Genetics, 2016. https://urn.nsk.hr/urn:nbn:hr:159:787761. ↩︎
Usserly, D. (1998): Gene Exchange, Meiosis & Eukaryotic Life Cycles. In Biology 101: Life on Planet Earth, http://www.cbs.dtu.dk/courses/genomics_course/roanoke/bio101ch12.htm. ↩︎
Eguizabal, C., N. Montserrat, R. Vassena, M. Barragan, E. Garreta, L. Garcia-Quevedo, F. Vidal, A. Giorgetti, A. Veiga, and J. C. Izpisua Belmonte. ‘Complete Meiosis from Human Induced Pluripotent Stem Cells’. STEM CELLS 29, no. 8 (2011): 1186–95. https://doi.org/10.1002/stem.672. ↩︎
Pierson Smela, Merrick, Jessica Adams, Carl Ma, Laura Breimann, Ursula Widocki, Toshi Shioda, and George M. Church. ‘Induction of Meiosis from Human Pluripotent Stem Cells’. bioRxiv, 31 May 2024, 2024.05.31.596483. https://doi.org/10.1101/2024.05.31.596483. ↩︎ ↩︎
Yuan, Yan, Laihua Li, Qing Cheng, Feiyang Diao, Qiao Zeng, Xiaoyu Yang, Yibo Wu, et al. ‘In Vitro Testicular Organogenesis from Human Fetal Gonads Produces Fertilization-Competent Spermatids’. Cell Research 30, no. 3 (March 2020): 244–55. https://doi.org/10.1038/s41422-020-0283-z. ↩︎ ↩︎
Hwang, Young Sun, Shinnosuke Suzuki, Yasunari Seita, Jumpei Ito, Yuka Sakata, Hirofumi Aso, Kei Sato, Brian P. Hermann, and Kotaro Sasaki. ‘Reconstitution of Prospermatogonial Specification in Vitro from Human Induced Pluripotent Stem Cells’. Nature Communications 11, no. 1 (9 November 2020): 5656. https://doi.org/10.1038/s41467-020-19350-3. ↩︎ ↩︎ ↩︎
Morohaku, Kanako, Ren Tanimoto, Keisuke Sasaki, Ryouka Kawahara-Miki, Tomohiro Kono, Katsuhiko Hayashi, Yuji Hirao, and Yayoi Obata. ‘Complete in Vitro Generation of Fertile Oocytes from Mouse Primordial Germ Cells’. Proceedings of the National Academy of Sciences of the United States of America 113, no. 32 (9 August 2016): 9021–26. https://doi.org/10.1073/pnas.1603817113. ↩︎
Hikabe, Orie, Nobuhiko Hamazaki, Go Nagamatsu, Yayoi Obata, Yuji Hirao, Norio Hamada, So Shimamoto, et al. ‘Reconstitution in Vitro of the Entire Cycle of the Mouse Female Germ Line’. Nature 539, no. 7628 (November 2016): 299–303. https://doi.org/10.1038/nature20104. ↩︎
Ron Slagter, O. Paul Gobée, LUMC, Hope Wicks, LUMC, et al. ‘Slagter - Drawing Human Oogenesis Diagram - English Labels | AnatomyTOOL’. Accessed 21 February 2025. https://anatomytool.org/content/slagter-drawing-human-oogenesis-diagram-english-labels. ↩︎
Garcia-Alonso, Luz, Valentina Lorenzi, Cecilia Icoresi Mazzeo, João Pedro Alves-Lopes, Kenny Roberts, Carmen Sancho-Serra, Justin Engelbert, et al. ‘Single-Cell Roadmap of Human Gonadal Development’. Nature 607, no. 7919 (July 2022): 540–47. https://doi.org/10.1038/s41586-022-04918-4. ↩︎
Mizuta, Ken, Yoshitaka Katou, Baku Nakakita, Aoi Kishine, Yoshiaki Nosaka, Saki Saito, Chizuru Iwatani, et al. ‘Ex Vivo Reconstitution of Fetal Oocyte Development in Humans and Cynomolgus Monkeys’. The EMBO Journal 41, no. 18 (15 September 2022): e110815. https://doi.org/10.15252/embj.2022110815. ↩︎
Baena, Valentina, and Mark Terasaki. ‘Three-Dimensional Organization of Transzonal Projections and Other Cytoplasmic Extensions in the Mouse Ovarian Follicle’. Scientific Reports 9, no. 1 (4 February 2019): 1262. https://doi.org/10.1038/s41598-018-37766-2. ↩︎
Mescher, Anthony L. ‘Junqueira’s Basic Histology: Text and Atlas, 17th Edition | AccessMedicine | McGraw Hill Medical’. Accessed 21 February 2025. https://accessmedicine.mhmedical.com/book.aspx?bookID=3390. ↩︎
Peters, H., A. G. Byskov, and J. Grinsted. ‘Follicular Growth in Fetal and Prepubertal Ovaries of Humans and Other Primates’. Clinics in Endocrinology and Metabolism 7, no. 3 (November 1978): 469–85. https://doi.org/10.1016/s0300-595x(78)80005-x. ↩︎
Anderson, R.A., M. McLaughlin, W.H.B. Wallace, D.F. Albertini, and E.E. Telfer. ‘The Immature Human Ovary Shows Loss of Abnormal Follicles and Increasing Follicle Developmental Competence through Childhood and Adolescence’. Human Reproduction (Oxford, England) 29, no. 1 (January 2014): 97–106. https://doi.org/10.1093/humrep/det388. ↩︎
Silber, Sherman J., Sierra Goldsmith, Leilani Castleman, and Katsuhiko Hayashi. ‘In Vitro Maturation, In Vitro Oogenesis, and Ovarian Longevity’. Reproductive Sciences 31, no. 5 (1 May 2024): 1234–45. https://doi.org/10.1007/s43032-023-01427-1. ↩︎ ↩︎
De Vos, Michel, Michaël Grynberg, Tuong M. Ho, Ye Yuan, David F. Albertini, and Robert B. Gilchrist. ‘Perspectives on the Development and Future of Oocyte IVM in Clinical Practice’. Journal of Assisted Reproduction and Genetics 38, no. 6 (June 2021): 1265–80. https://doi.org/10.1007/s10815-021-02263-5. ↩︎
Kawamura, Kazuhiro, Yuan Cheng, Nao Suzuki, Masashi Deguchi, Yorino Sato, Seido Takae, Chi-hong Ho, et al. ‘Hippo Signaling Disruption and Akt Stimulation of Ovarian Follicles for Infertility Treatment’. Proceedings of the National Academy of Sciences 110, no. 43 (22 October 2013): 17474–79. https://doi.org/10.1073/pnas.1312830110. ↩︎ ↩︎
Hatırnaz, Şafak, Barış Ata, Ebru Saynur Hatırnaz, Michael Haim Dahan, Samer Tannus, Justin Tan, and Seang Lin Tan. ‘Oocyte in Vitro Maturation: A Sytematic Review’. Turkish Journal of Obstetrics and Gynecology 15, no. 2 (June 2018): 112–25. https://doi.org/10.4274/tjod.23911. ↩︎ ↩︎
Guglielmo, Maria Cristina, and David F. Albertini. ‘The Structural Basis for Coordinating Oogenesis and Folliculogenesis’. In Oogenesis, edited by Giovanni Coticchio, David F. Albertini, and Lucia De Santis, 63–73. London: Springer, 2013. https://doi.org/10.1007/978-0-85729-826-3_5. ↩︎ ↩︎
Malo, Clara, Sara Oliván, Ignacio Ochoa, and Ariella Shikanov. ‘In Vitro Growth of Human Follicles: Current and Future Perspectives’. International Journal of Molecular Sciences 25, no. 3 (26 January 2024): 1510. https://doi.org/10.3390/ijms25031510. ↩︎
Pierson Smela, Merrick D, Christian C Kramme, Patrick RJ Fortuna, Jessica L Adams, Rui Su, Edward Dong, Mutsumi Kobayashi, et al. ‘Directed Differentiation of Human iPSCs to Functional Ovarian Granulosa-like Cells via Transcription Factor Overexpression’. Edited by T Rajendra Kumar and Marianne E Bronner. eLife 12 (21 February 2023): e83291. https://doi.org/10.7554/eLife.83291. ↩︎
Yoshino, Takashi, Takahiro Suzuki, Go Nagamatsu, Haruka Yabukami, Mika Ikegaya, Mami Kishima, Haruka Kita, et al. ‘Generation of Ovarian Follicles from Mouse Pluripotent Stem Cells’. Science (New York, N.Y.) 373, no. 6552 (16 July 2021): eabe0237. https://doi.org/10.1126/science.abe0237. ↩︎
Ron Slagter, O. Paul Gobée, LUMC, Hope Wicks, LUMC, et al. ‘Slagter - Drawing Human Oogenesis Diagram - English Labels | AnatomyTOOL’. Accessed 23 February 2025. https://anatomytool.org/content/slagter-drawing-human-spermatogenesis-diagram-english-labels. ↩︎
Gilbert, Scott F. ‘Spermatogenesis’. In Developmental Biology. 6th Edition. Sinauer Associates, 2000. https://www.ncbi.nlm.nih.gov/books/NBK10095/. ↩︎
Sato, Takuya, Kumiko Katagiri, Ayako Gohbara, Kimiko Inoue, Narumi Ogonuki, Atsuo Ogura, Yoshinobu Kubota, and Takehiko Ogawa. ‘In Vitro Production of Functional Sperm in Cultured Neonatal Mouse Testes’. Nature 471, no. 7339 (24 March 2011): 504–7. https://doi.org/10.1038/nature09850. ↩︎
Matsumura, Takafumi, Kumiko Katagiri, Tatsuma Yao, Yu Ishikawa-Yamauchi, Shino Nagata, Kiyoshi Hashimoto, Takuya Sato, et al. ‘Generation of Rat Offspring Using Spermatids Produced through in Vitro Spermatogenesis’. Scientific Reports 13, no. 1 (26 July 2023): 12105. https://doi.org/10.1038/s41598-023-39304-1. ↩︎
Ishikura, Yukiko, Hiroshi Ohta, Takuya Sato, Yusuke Murase, Yukihiro Yabuta, Yoji Kojima, Chika Yamashiro, et al. ‘In Vitro Reconstitution of the Whole Male Germ-Cell Development from Mouse Pluripotent Stem Cells’. Cell Stem Cell 28, no. 12 (2 December 2021): 2167-2179.e9. https://doi.org/10.1016/j.stem.2021.08.005. ↩︎
Whelan, Eoin C., Young Sun Hwang, Yasunari Seita, Ryo Yokomizo, N. Adrian Leu, Keren Cheng, and Kotaro Sasaki. ‘Generation of Spermatogonia from Pluripotent Stem Cells in Humans and Non-Human Primates’. bioRxiv, 6 May 2024. https://doi.org/10.1101/2024.05.03.592203. ↩︎ ↩︎
Chen, Haiqi, Evan Murray, Anubhav Sinha, Anisha Laumas, Jilong Li, Daniel Lesman, Xichen Nie, et al. ‘Dissecting Mammalian Spermatogenesis Using Spatial Transcriptomics’. Cell Reports 37, no. 5 (2 November 2021). https://doi.org/10.1016/j.celrep.2021.109915. ↩︎
Sosa, Enrique, Di Chen, Ernesto J. Rojas, Jon D. Hennebold, Karen A. Peters, Zhuang Wu, Truong N. Lam, et al. ‘Differentiation of Primate Primordial Germ Cell-like Cells Following Transplantation into the Adult Gonadal Niche’. Nature Communications 9 (17 December 2018): 5339. https://doi.org/10.1038/s41467-018-07740-7. ↩︎
Cheng, Hanhua, Dantong Shang, and Rongjia Zhou. ‘Germline Stem Cells in Human’. Signal Transduction and Targeted Therapy 7, no. 1 (2 October 2022): 1–22. https://doi.org/10.1038/s41392-022-01197-3. ↩︎
Ibtisham, Fahar, and Ali Honaramooz. ‘Spermatogonial Stem Cells for In Vitro Spermatogenesis and In Vivo Restoration of Fertility’. Cells 9, no. 3 (March 2020): 745. https://doi.org/10.3390/cells9030745. ↩︎
Kulibin, A. Yu, and E. A. Malolina. ‘In Vitro Spermatogenesis: In Search of Fully Defined Conditions’. Frontiers in Cell and Developmental Biology 11 (24 February 2023). https://doi.org/10.3389/fcell.2023.1106111. ↩︎
MacCarthy, Caitlin M., Guangming Wu, Vikas Malik, Yotam Menuchin-Lasowski, Taras Velychko, Gal Keshet, Rui Fan, et al. ‘Highly Cooperative Chimeric Super-SOX Induces Naive Pluripotency across Species’. Cell Stem Cell 31, no. 1 (4 January 2024): 127-147.e9. https://doi.org/10.1016/j.stem.2023.11.010. ↩︎
Meng, Li, John J. Ely, Richard L. Stouffer, and Don P. Wolf. ‘Rhesus Monkeys Produced by Nuclear Transfer1’. Biology of Reproduction 57, no. 2 (1 August 1997): 454–59. https://doi.org/10.1095/biolreprod57.2.454. ↩︎
Bayerl, Jonathan, Muneef Ayyash, Tom Shani, Yair Shlomo Manor, Ohad Gafni, Rada Massarwa, Yael Kalma, et al. ‘Principles of Signaling Pathway Modulation for Enhancing Human Naive Pluripotency Induction’. Cell Stem Cell 28, no. 9 (2 September 2021): 1549-1565.e12. https://doi.org/10.1016/j.stem.2021.04.001. ↩︎
Tan, Kun, Hye-Won Song, Merlin Thompson, Sarah Munyoki, Meena Sukhwani, Tung-Chin Hsieh, Kyle E. Orwig, and Miles F. Wilkinson. ‘Transcriptome Profiling Reveals Signaling Conditions Dictating Human Spermatogonia Fate in Vitro’. Proceedings of the National Academy of Sciences 117, no. 30 (28 July 2020): 17832–41. https://doi.org/10.1073/pnas.2000362117. ↩︎
Fischer, Laura A., Brittany Meyer, Monica Reyes, Joseph E. Zemke, Jessica K. Harrison, Kyoung-mi Park, Ting Wang, Harald Jüppner, Sabine Dietmann, and Thorold W. Theunissen. ‘Tracking and Mitigating Imprint Erasure during Induction of Naive Human Pluripotency at Single-Cell Resolution’. Stem Cell Reports 0, no. 0 (13 February 2025). https://doi.org/10.1016/j.stemcr.2025.102419. ↩︎
Nakamura, Muneaki, Yuchen Gao, Antonia A. Dominguez, and Lei S. Qi. ‘CRISPR Technologies for Precise Epigenome Editing’. Nature Cell Biology 23, no. 1 (January 2021): 11–22. https://doi.org/10.1038/s41556-020-00620-7. ↩︎
McCutcheon, Sean R., Dahlia Rohm, Nahid Iglesias, and Charles A. Gersbach. ‘Epigenome Editing Technologies for Discovery and Medicine’. Nature Biotechnology 42, no. 8 (August 2024): 1199–1217. https://doi.org/10.1038/s41587-024-02320-1. ↩︎
Liesenfelder, Sven, Mohamed H. Elsafi Mabrouk, Jessica Iliescu, Monica Varona Baranda, Athanasia Mizi, Martina Wessiepe, Argyris Papantonis, and Wolfgang Wagner. ‘Epigenetic Editing at Individual Age-Associated CpGs Affects the Genome-Wide Epigenetic Aging Landscape’. bioRxiv, 5 June 2024. https://doi.org/10.1101/2024.06.04.597161. ↩︎
Dechiara, Thomas M., William T. Poueymirou, Wojtek Auerbach, David Frendewey, George D. Yancopoulos, and David M. Valenzuela. ‘VelociMouse: Fully ES Cell-Derived F0-Generation Mice Obtained from the Injection of ES Cells into Eight-Cell-Stage Embryos’. Methods in Molecular Biology (Clifton, N.J.) 530 (2009): 311–24. https://doi.org/10.1007/978-1-59745-471-1_16. ↩︎
Poirot, Catherine, Anne Fortin, Nathalie Dhédin, Pauline Brice, Gérard Socié, Jean-Marc Lacorte, Jean-Paul Akakpo, et al. ‘Post-Transplant Outcome of Ovarian Tissue Cryopreserved after Chemotherapy in Hematologic Malignancies’. Haematologica 104, no. 8 (August 2019): e360–63. https://doi.org/10.3324/haematol.2018.211094. ↩︎
Nikiforov, Dmitry, Cheng Junping, Jesus Cadenas, Vallari Shukla, Robert Blanshard, Susanne Elisabeth Pors, Stine Gry Kristensen, et al. ‘Improving the Maturation Rate of Human Oocytes Collected Ex Vivo during the Cryopreservation of Ovarian Tissue’. Journal of Assisted Reproduction and Genetics 37, no. 4 (1 April 2020): 891–904. https://doi.org/10.1007/s10815-020-01724-7. ↩︎
Benson-Tilsen, Tsvi. ‘The Power of Selection’, 9 August 2022. https://tsvibt.blogspot.com/2022/08/the-power-of-selection.html. ↩︎ ↩︎
Bell, Avery Davis, Curtis J. Mello, James Nemesh, Sara A. Brumbaugh, Alec Wysoker, and Steven A. McCarroll. ‘Insights into Variation in Meiosis from 31,228 Human Sperm Genomes’. Nature 583, no. 7815 (July 2020): 259–64. https://doi.org/10.1038/s41586-020-2347-0. ↩︎ ↩︎ ↩︎ ↩︎ ↩︎
Doležel, Jaroslav, Sergio Lucretti, István Molnár, Petr Cápal, and Debora Giorgi. ‘Chromosome Analysis and Sorting’. Cytometry Part A 99, no. 4 (2021): 328–42. https://doi.org/10.1002/cyto.a.24324. ↩︎
Chankitisakul, Vibuntita, Nutthee Am-In, Theerawat Tharasanit, Tamas Somfai, Takashi Nagai, and Mongkol Techakumphu. ‘Sperm Pretreatment with Dithiothreitol Increases Male Pronucleus Formation Rates After Intracytoplasmic Sperm Injection (ICSI) in Swamp Buffalo Oocytes’. Journal of Reproduction and Development 59, no. 1 (2013): 66–71. https://doi.org/10.1262/jrd.2012-104. ↩︎ ↩︎
Frade, João, Shoma Nakagawa, Paola Cortes, Umberto di Vicino, Neus Romo, Frederic Lluis, and Maria Pia Cosma. ‘Controlled Ploidy Reduction of Pluripotent 4n Cells Generates 2n Cells during Mouse Embryo Development’. Science Advances 5, no. 10 (16 October 2019): eaax4199. https://doi.org/10.1126/sciadv.aax4199. ↩︎
Gwern Branwen. ‘History of Iterated Embryo Selection’, 18 January 2019. https://gwern.net/ies-history. ↩︎
Metacelsus. ‘Meiosis Is All You Need’. Substack newsletter. De Novo (blog), 30 June 2022. https://denovo.substack.com/p/meiosis-is-all-you-need. ↩︎
Trontelj, Katja, Marko Ušaj, and Damijan Miklavčič. ‘Cell Electrofusion Visualized with Fluorescence Microscopy’. Journal of Visualized Experiments : JoVE, no. 41 (1 July 2010): 1991. https://doi.org/10.3791/1991. ↩︎
Sagi, Ido, Gloryn Chia, Tamar Golan-Lev, Mordecai Peretz, Uri Weissbein, Lina Sui, Mark V. Sauer, Ofra Yanuka, Dieter Egli, and Nissim Benvenisty. ‘Derivation and Differentiation of Haploid Human Embryonic Stem Cells’. Nature 532, no. 7597 (April 2016): 107–11. https://doi.org/10.1038/nature17408. ↩︎
Tsui, Vanessa, Ruqian Lyu, Stevan Novakovic, Jessica M. Stringer, Jessica E.M. Dunleavy, Elissah Granger, Tim Semple, et al. ‘Fancm Has Dual Roles in the Limiting of Meiotic Crossovers and Germ Cell Maintenance in Mammals’. Cell Genomics 3, no. 8 (August 2023): 100349. https://doi.org/10.1016/j.xgen.2023.100349. ↩︎
Tesarik, Jan, Mustafa Bahceci, Cenk Özcan, Ermanno Greco, and Carmen Mendoza. ‘Restoration of Fertility by In-Vitro Spermatogenesis’. The Lancet 353, no. 9152 (13 February 1999): 555–56. https://doi.org/10.1016/S0140-6736(98)04784-9. ↩︎
Cremades, N., R. Bernabeu, A. Barros, and M. Sousa. ‘In-Vitro Maturation of Round Spermatids Using Co-Culture on Vero Cells’. Human Reproduction (Oxford, England) 14, no. 5 (May 1999): 1287–93. https://doi.org/10.1093/humrep/14.5.1287. ↩︎
Tanaka, Atsushi, Motoki Nagayoshi, Shoichiro Awata, Yoshifumi Mawatari, Izumi Tanaka, and Hiroshi Kusunoki. ‘Completion of Meiosis in Human Primary Spermatocytes through in Vitro Coculture with Vero Cells’. Fertility and Sterility 79 (1 March 2003): 795–801. https://doi.org/10.1016/S0015-0282(02)04833-1. ↩︎
Tanaka, Atsushi, Motoi Nagayoshi, Shoichiro Awata, Izumi Tanaka, and Hiroshi Kusunoki. ‘Differentiation of Human Round Spermatids into Motile Spermatozoa through in Vitro Coculture with Vero Cells’. Reproductive Medicine and Biology 8, no. 4 (21 August 2009): 169–75. https://doi.org/10.1007/s12522-009-0030-0. ↩︎
Easley, Charles A., Bart T. Phillips, Megan M. McGuire, Jennifer M. Barringer, Hanna Valli, Brian P. Hermann, Calvin R. Simerly, et al. ‘Direct Differentiation of Human Pluripotent Stem Cells into Haploid Spermatogenic Cells’. Cell Reports 2, no. 3 (27 September 2012): 440–46. https://doi.org/10.1016/j.celrep.2012.07.015. ↩︎
Perrard, Marie-Hélène, Nicolas Sereni, Caroline Schluth-Bolard, Antonine Blondet, Sandrine Giscard d′Estaing, Ingrid Plotton, Nicolas Morel-Journel, Hervé Lejeune, Laurent David, and Philippe Durand. ‘Complete Human and Rat Ex Vivo Spermatogenesis from Fresh or Frozen Testicular Tissue1’. Biology of Reproduction 95, no. 4 (1 October 2016): 89, 1–10. https://doi.org/10.1095/biolreprod.116.142802. ↩︎
Michele, Francesca de, Jonathan Poels, Maxime Vermeulen, Jérôme Ambroise, Damien Gruson, Yves Guiot, and Christine Wyns. ‘Haploid Germ Cells Generated in Organotypic Culture of Testicular Tissue From Prepubertal Boys’. Frontiers in Physiology 9 (9 October 2018). https://doi.org/10.3389/fphys.2018.01413. ↩︎
Sun, Min, Qingqing Yuan, Minghui Niu, Hong Wang, Liping Wen, Chencheng Yao, Jingmei Hou, et al. ‘Efficient Generation of Functional Haploid Spermatids from Human Germline Stem Cells by Three-Dimensional-Induced System’. Cell Death & Differentiation 25, no. 4 (April 2018): 749–66. https://doi.org/10.1038/s41418-017-0015-1. ↩︎
Gallegos, Maria. Fantastic Genes and Where to Find Them. Updated 2022-09-13. Accessed 16 February 2025. https://bookdown.org/maria_gallegos/where-are-genes/#preface. ↩︎ ↩︎
Chen, Jian-Min, David N. Cooper, Nadia Chuzhanova, Claude Férec, and George P. Patrinos. ‘Gene Conversion: Mechanisms, Evolution and Human Disease’. Nature Reviews Genetics 8, no. 10 (October 2007): 762–75. https://doi.org/10.1038/nrg2193. ↩︎
Perman, Mihael, and Jon A. Wellner. ‘An Excursion Approach to Maxima of the Brownian Bridge’. Stochastic Processes and Their Applications 124, no. 9 (1 September 2014): 3106–20. https://doi.org/10.1016/j.spa.2014.04.008. ↩︎
Xiao, Felix. ‘The Brownian Bridge Joint Max Position Distribution’. Undersampled - Felix Xiao, 24 January 2018. https://felixxiao.github.io/2018/01/brownian-bridge. ↩︎
Ottolini, Christian S., Louise Newnham, Antonio Capalbo, Senthilkumar A. Natesan, Hrishikesh A. Joshi, Danilo Cimadomo, Darren K. Griffin, et al. ‘“Genome-Wide Recombination and Chromosome Segregation in Human Oocytes and Embryos Reveal Selection for Maternal Recombination Rates”’. Nature Genetics 47, no. 7 (July 2015): 727–35. https://doi.org/10.1038/ng.3306. ↩︎
Palamara, Pier Francesco, Laurent C. Francioli, Peter R. Wilton, Giulio Genovese, Alexander Gusev, Hilary K. Finucane, Sriram Sankararaman, et al. ‘Leveraging Distant Relatedness to Quantify Human Mutation and Gene-Conversion Rates’. The American Journal of Human Genetics 97, no. 6 (December 2015): 775–89. https://doi.org/10.1016/j.ajhg.2015.10.006. ↩︎
Savage, Jeanne E, Philip R Jansen, Sven Stringer, Kyoko Watanabe, Julien Bryois, Christiaan A de Leeuw, Mats Nagel, et al. ‘Genome-Wide Association Meta-Analysis in 269,867 Individuals Identifies New Genetic and Functional Links to Intelligence’. Nature Genetics 50, no. 7 (July 2018): 912–19. https://doi.org/10.1038/s41588-018-0152-6. ↩︎
28 comments
Comments sorted by top scores.
comment by Metacelsus · 2025-03-09T16:06:15.570Z · LW(p) · GW(p)
I don't know if targeted crossover is plausibly feasible.
It is definitely feasible. This is how artificial gene drives work.
comment by Metacelsus · 2025-03-09T15:53:12.070Z · LW(p) · GW(p)
So that's why you can't just make a human baby without knowing what you're doing: You stand a high risk of making a baby with developmental abnormalities that weren't severe enough to abort the fetus, but are severe enough that the child is suffering. If for some reason the moral consequences of that aren't enough to dissuade you, consider that other people would ban you and your children and your children's children and your artificial children and any similar research for 1000 years.
I cannot overemphasize this!
comment by solhando · 2025-03-04T16:29:56.804Z · LW(p) · GW(p)
I'm a layman, but the "Hulk Sperm" method seems the most plausible EC-bypass method to me, and EC-bypass generally seems a more plausible route than EC-making. After reading the paper, it seems like any of the routes to EC-making seem either speculative, or very prone to failure (while also being hard to verify as a failure).
an issue with spermatogonial transplant into testicles is that GV (e.g. edited) spermatogonia are likely to be stressed and imperfectly maintained, and therefore are likely to be outcompeted by normal spermatogonia and die out
If the problem is a concern with competitiveness after re-transplantation, is there a way to create an environment where the stressed spermatogonia has either reduced competition, or is more competitive relative to its unedited counterparts?
Off the top of my head this might look like locally "sterilizing" just the spermatogonia tissue after a biopsy somehow, without damaging the machinery that leads to functional spermatozoon development and paternal imprinting. I'm not sure if this sort of targeted wiping is possible though, but at least with two testicles, the option of sterilizing one from spermatogonia tissue wouldn't necessarily lead to complete sterilization. Or even if our only option here was complete spermatogonia sterilization, sperm preservation is relatively easy, so it still might be tolerable.
Alternatively, rather than just editing in a fluorescent reporter, you might also edit in a gene that makes the edited sperm more competitive in an environment that can be artificially induced in vivo. With just a fluorescent reporter, such a change arising naturally would naturally lead to the sperm being outcompeted and dying out (or if it as a completely neutral adaptation maybe not?), but if we induce conditions that are more favorable to spermatogonia with a specific tolerance induced by an inserted gene, they may have enough of a competitive advantage to persist in meaningful numbers. Off the top of my head I would consider higher temperature tolerance (with higher temperatures induced with a willy-warmer or something), or perhaps resistance to a chemical that's normally harmful to spermatozoon? No idea what this could be though.
Also, would a fluorescent reporter gene edited into spermatogonia and present in spermatozoon persist into the zygote, and therefore into all the descended cells of the eventual mature organism? I think it would be hilarious and incredibly fitting if a side effect of making supermen is that they literally glow.
Replies from: TsviBT↑ comment by TsviBT · 2025-03-04T22:33:28.108Z · LW(p) · GW(p)
"Hulk Sperm" method seems the most plausible EC-bypass method to me, and EC-bypass generally seems a more plausible route than EC-making
I partly agree, though with qualifications. One thing to note is that SSC methods are strictly haploid, hence half the power of diploid methods. Another thing to note is that SSC methods, especially ones that require iteration (editing), are not trivial in terms of EC; you have to maintain imprints, probably, as well as maintaining the general stem cell niche (i.e. ability to proliferate and ability to differentiate into spermatocytes).
I find e.g. sperm chromosome to have a comparable chance of working soon, though it has its own somewhat separate challenges.
Here "plausible" means "plausible really soon, like in a couple years". The more robust way, that will quite likely work eventually, is more natural gametogenesis / EC-making (IVO and IVS). These have far more investment from scientists, so in particular it's somewhat more clear what it would take and how to accelerate it, though far from fully clear. But yes, they do seem harder.
Yes, sterilization is possible; they sterilize mouse testes before retransplanting exogenous SSCs.
Sterilizing one testis is an interesting suggestion I hadn't thought of, thanks. It does seem like a big cost, but presumably much less than half the cost of sterilizing both. It's still a cost because you're decreasing your fertility, and because IDK if there are sterilization methods that aren't more generally toxic (e.g. oncogenic or something).
locally "sterilizing" just the spermatogonia tissue after a biopsy somehow, without damaging the machinery that leads to functional spermatozoon development and paternal imprinting. I'm not sure if this sort of targeted wiping is possible though,
Yeah, IDK either, interesting question. I would very weakly guess that native SSCs can colonize the whole testis but IDK.
you might also edit in a gene that makes the edited sperm more competitive in an environment that can be artificially induced in vivo
Yeah, this is a good idea that I've heard in the abstract. I don't know about implementation details. I think I get pretty nervous whenever we're talking about some unnatural modification, i.e. some modification that isn't already present in natural humans. The case for safety seems significantly harder to make. If we do adult somatic gene therapies that are not just modifying to a common allele, and it's fine, that could be some evidence that it's fine. But SSCs in particular are stem cells, so a priori they are cancer risks, I would think. Like "how can I make this cell proliferate more than natural, and outcompete its natural healthy counterparts" is kinda like asking "how can I make some cancer".
would a fluorescent reporter gene edited into spermatogonia and present in spermatozoon persist into the zygote, and therefore into all the descended cells of the eventual mature organism?
The idea is that you put the reporter near a gene whose protein is only present in sperm--e.g. a meiosis-specific protein (which I have heard do exist). So the child wouldn't glow. But their gametes, and their descendants's gametes (with 50% probability, etc.) would glow.
comment by DaemonicSigil · 2025-03-04T07:45:05.950Z · LW(p) · GW(p)
Re the "Appendix: Cheap DNA segment sensing" section, just going to throw out a thought that occurred to me (very much a non-expert). Let's say we're doing IVS, and assume we can separate spermatocytes into separate microwells before they undergo meiosis. The starting cells all have a known genome. Then the cell in each microwell divides into 4 cells. If we sequence 3 of them, then we know by process of elimination what the sequence on the 4th cell is, at a very high level of detail, including crossovers, etc. So we kill 3 cells and look at their DNA, and then we know what DNA the remaining living cell has without doing anything to it.
Okay, DNA sequencing is still fairly expensive, so maybe it's super crazy to do it 3 times to get a single cell with known DNA. But:
- Maybe sequencing will get cheaper.
- The same trick should work for existing cheap methods that give coarser information. Eg. one can freely decondense the sperm DNA for FISH, without worrying about damaging the cell, because it's one of the 3 that's going to die anyway.
If it's too hard to separate the cells into microwells while they're still dividing, maybe there are alternate things we could do like just watching the culture with a microscope and keeping track of who split from who and where they ended up (plus some kind of microfluidics setup to shuffle the sperms around to where we want them).
Replies from: TsviBT, TsviBT↑ comment by TsviBT · 2025-03-04T07:59:45.652Z · LW(p) · GW(p)
Yep, this is the standard first "clever person starts thinking about nondestructive sequencing" thing. E.g. I wrote about it here: https://tsvibt.blogspot.com/2022/06/non-destructively-sequencing-gametes-by.html and Gwern mentions it. It's addressed in the present article here:
In theory one could do "entanglement sequencing", wherein you capture the four meiotic "grandchildren" of a single gametogonium (progenitor stem cell that produces gametes). This is impractical as far as I know, because both oogenesis and spermatogenesis are complex and all but require a bunch of surrounding tissue. Spermatogenesis in particular involves a complicated process where many partially-differentiated progenitor cells stay tightly connected as they pass through the seminiferous tubules. Oocytes might be more feasible to indirectly sequence, e.g. by looking at the polar body emitted after false fertilization.
You're talking about if we do have IVS. In that case, yes, plausibly you could do it. But this wouldn't enable recombinant chromosome selection unless you have a way of sensing index; which is plausibly easier, but IDK how much easier. And anyway, the hardest part of CS is chromosome transplantation.
It would enable simple gamete selection. But that kinda sucks.
If you have full IVS, there's almost certainly more powerful things you can do; if nothing else, you can do a ton of CRISPR editing of cultured iPSCs, and then IVS.
Replies from: DaemonicSigil↑ comment by DaemonicSigil · 2025-03-04T21:06:45.608Z · LW(p) · GW(p)
Thanks for the reply & link. I definitely missed that paragraph, whoops.
IMO even just simple gamete selection would be pretty great for avoiding the worst genetic diseases. I guess tracking nuclei with a microscope is way more feasible than the microwell thing, given how hard it looks to make IVS work at all.
Replies from: TsviBT↑ comment by TsviBT · 2025-03-04T22:43:49.071Z · LW(p) · GW(p)
I think the numbers just kind of suck. I didn't go into them much because gamete selection seems largely hypothetical. Like, the procedure here seems kinda expensive per-spermatocyte (guy who divides into 4 sperm). I gesture at how to compute it here: https://www.lesswrong.com/posts/2w6hjptanQ3cDyDw7/methods-for-strong-human-germline-engineering#The_power_of_single_gamete_selection [LW · GW]
To give some numbers, consider 10k sperm. We look in the embryo selection powers:
For 10k, i.e. e4.0, you get about 2.7 raw SDs, for embros; for sperm you multiply by the sperm SD scale, which is 1/√2 because sperm have half the variance of embryos. That's about 1.9 SDs. Then you do a round of embryo selection, with say 10 embryos, for about .7 more raw SDs (because you're mainly selecting on the maternal DNA). Total is 2.6 raw SDs. As IQ points this is like 13 IQ points. It's pretty interesting, but not groundbreaking; and even with 10k sperm, could actually be pretty expensive, like would plausibly be at least several $100k. Another order of magnitude sperm would be another OOM expensiver, and only a very small bump in power. I haven't run the numbers for disease reduction, and yeah it would probably be pretty nontrivial (because the marginal returns for diseases are high at the beginning, starting at a normal genome), but yeah it also does not qualify as strong GV.
To be clear, this would be awesome and someone should totally try, and it could plausibly synergize with other stuff. It's just not a priority for getting to strong GV.
↑ comment by TsviBT · 2025-03-27T20:38:55.890Z · LW(p) · GW(p)
(BTW I think you asking about entanglement sequencing caused me to a few days later realize that for chromosome selection, you can do at least index sensing by taking 1 chromosome randomly from a cell, and then sequencing/staining the remaining 22 (or 45), and seeing which index is missing. So thanks :) )
comment by samuelshadrach (xpostah) · 2025-03-03T21:28:13.138Z · LW(p) · GW(p)
I haven't made up my mind on whether I endorse human genetic engg, but I have technical doubts:
1. For simple embryonic selection, shouldn't we consider highest IQ of male embryos rather expected IQ of the embryos?
If I understand correctly, there is a bottleneck on eggs per egg donor, but not as tight a bottleneck on sperm cells per sperm donor. Assume there are 10,000 egg donors with high IQ, 100 eggs per donor mating with 1,000,000 sperm of one sperm donor with high IQ. Out of the 1,000,000 embryos, let's say the highest IQ embryo grows to childbearing age and is then mated with 100 eggs each of 10k egg donors in the second generation. IQ gain per generation will now be measured as M(10^6)/1.414, which is missing from your table.
2. How do you reason about theoretical IQ that falls outside the range where IQ tests have validity?
Assume my point 1 on simple selection is correct and +3 SD IQ gain per generation is achievable. Assume we start with +3 SD IQ donors. After two generations that's already a +9 SD IQ adult male. How do you make sense of a number like +9 SD IQ?
3. Is age 18 for sperm and egg donation a legal limit or a biological one?
I'm asking because if it is not a biological limit, I can imagine a govt changing the law for various reasons, for example in order to win an arms race against another country.
When we calculate IQ gain per generation, is this IQ gain per 18 years or IQ gain per 12 years?
Replies from: TsviBT, TsviBT↑ comment by TsviBT · 2025-03-03T22:40:10.150Z · LW(p) · GW(p)
How do you make sense of a number like +9 SD IQ?
Yeah, I don't know if it makes much sense, and haven't thought too much about it. A few points:
-
I don't know if I actually care too much. Or rather, I think it would be awesome if +9 SD IQ just makes sense somehow, and also we can enable parents to choose that, and also it flows into more generally being insightful. But I think just having tons of people sample from a distribution with a mean of +6 SDs already makes the outlook for humanity significantly better on my view. It's not remotely the case that every such person will be a great scientist or philosopher; people will have different interests, we shouldn't be strongly conscripting GE children, and there are likely several other traits quite important for contributing to defending against humanity's greatest threats (e.g. curiosity, bravery, freethink; attention, determination, drive; wisdom [LW · GW], reflectiveness).
-
Actually targeting +9 SDs on anything, especially IQ, is potentially quite dangerous and should either be done with great caution after further study, or not at all. See the bullet point "Traits outside the regime of adaptedness".
-
But if I speculate:
- Some genetic variants will be about "sand in the gears" of the brain. It doesn't seem crazy to think that you can get a lot of performance by just removing an exceptionally large amount of this. But IDK how much there actually is; kman might have suggested that this isn't actually much of the genetic variance in IQ.
- Some genetic variants will be about "scaling up" (e.g. literally growing a bigger brain, or a more vascularized one, or one with a higher metabolic budget to spend on plasticity, or something like that, IDK). These seem like they plausibly could keep having meaningful effects past the human envelope, but on the other hand could easily hit limits discussed in "Traits outside...".
- Some genetic variants will be about, vaguely, budgeting resources between different neural algorithms. These could easily keep having effects outside the human envelope (e.g., let's have AN EVEN BIGGER MATH BRAIN CHUNK EVEN THAN EINSTEIN, or what have you). On the other hand, you could very plausibly overshoot, and get some weird or dysfunctional result.
- Cf. jimrandomh's comment about hyperparameters and overshooting here: https://www.lesswrong.com/posts/DfrSZaf3JC8vJdbZL/how-to-make-superbabies?commentId=C7MvCZHbFmeLdxyAk [LW(p) · GW(p)]
↑ comment by samuelshadrach (xpostah) · 2025-03-04T06:56:51.965Z · LW(p) · GW(p)
Got it.
On a technical level, I think more speculating is good before we run the experiment, given that these people if born may very well end up the most powerful people in history. Even small differences in the details could lead to very different futures for humanity.
On a non-technical level, it might be worth writing a post about your stance on the morality and politics of this. So we can separate that whole discussion from the technical discussion.
Replies from: TsviBT↑ comment by TsviBT · 2025-03-04T07:05:23.913Z · LW(p) · GW(p)
Even small differences in the details could lead to very different futures for humanity.
I don't super agree with this. But also, I'd view that as somewhat of a failure. Part of why I want the technology to be wideley available is that I think that decreases the path-dependence. Lots of diverse GE kids means a more universalist future, hopefully.
it might be worth writing a post about your stance on the morality and politics of this.
Yeah. I have several articles I want to write, though IDK which will become high-priority enough. Some thoughts on genomic liberty are here: https://www.lesswrong.com/posts/DfrSZaf3JC8vJdbZL/how-to-make-superbabies?commentId=ZeranH3yDBGWNxZ7h [LW(p) · GW(p)]
Replies from: xpostah↑ comment by samuelshadrach (xpostah) · 2025-03-04T07:45:37.957Z · LW(p) · GW(p)
Cool!
Have you read meditations on moloch?
My view on this is that even when individuals and countries are not under tight “adapt or die” competition constraints such as during wartime or poverty, everyone faces incentive gradients. Free choices aren’t exactly free. For instance I was “free” to not learn software development and pick a lower paying job, but someone from the outside could still predict with high likelihood I was going to learn software anyway.
Replies from: TsviBT↑ comment by TsviBT · 2025-03-04T08:15:25.273Z · LW(p) · GW(p)
I have read it (long ago).
I take the general point that making this technology partially removes a barrier, where previously human influence on children is limited, and afterward there is at least somewhat more influence. E.g. this could lead to:
- Sacrificing wellbeing for competitiveness
- Social pressure / "soft eugenics"
- Competitive selection (where I mentioned the Meditations)
One point of comparison is the default. There is a human-evolution that is always happening. Do we like its results? Do we trust it?
Another thing to point out is that the barrier is only somewhat eroded. Except for whole genome synthesis, the amount of control that germline engineering is fairly small compared to the total genetic and phenotypic variation in humans. You and I have 5 or 10 million differing alleles between us; GV would have an effect that's comparable to, say, 1000s of alleles. (This doesn't directly make sense for selection, but morally speaking.) In terms of phenotypes, most of the variation would still be in uncontrolled genomic differences and non-genetic differences. Current IQ PGSes explain <20% of the variance in IQ. Now, to some extent I can't have it both ways; either the benefits of GE are enormous because we're controlling traits somewhat, or we aren't controlling traits much and the benefits can't be that big. But think, for example, of shifting the mean of your kid's expected IQ, without much shifting the variance. (For some disease traits you can drive the probability of the disease far down, which is a lot of phenotypic control; but that seems not so bad!)
Replies from: xpostah↑ comment by samuelshadrach (xpostah) · 2025-03-04T09:55:19.852Z · LW(p) · GW(p)
I’m glad you’re thinking about it.
I would still encourage you to forecast what capabilities look like not just as of 2025, but after a trillion dollars of R&D enter this space. Mobilising a trillion dollars for a field of such importance is not difficult, once successful clinical results are out. All your claims about mean and variance, or about whole genome synthesis being possible, will no longer apply afaik.
↑ comment by TsviBT · 2025-03-03T22:45:27.684Z · LW(p) · GW(p)
For simple embryonic selection, shouldn't we consider highest IQ of male embryos rather expected IQ of the embryos?
There's something really off about the frame of your question. I'm not exactly sure where you're coming from. I'm not trying to direct anyone's reproduction, I'm not trying to influence anything at a population level, and also I'm not really focused on anything about multiple generations.
Replies from: xpostah↑ comment by samuelshadrach (xpostah) · 2025-03-04T06:40:03.539Z · LW(p) · GW(p)
Hmm
So I get that you want to do things with the consent of everyone involved, be it the sperm donor, egg donor, or the people who will actually raise the child. This doesn’t preclude thinking about population-level changes or thinking ahead multi-generational consequences.
Even if people don’t explicitly aim for population changes, these might be the emergent effects. It may be individually rational for each person to find the highest trait sperm donors they can find, even if they haven’t all coordinated with each other to do it.
More important though, your noble intentions are going to matter only so much once this tech is the public domain. People of various types of intentions may have access to this. If we have anything resembling markets + democracy + internet + multipolar nuclear world, a maxim that makes some sense to me is “don’t release any tech unless you’re ready for your worst enemies to have access to it as well”. You should assume, for example, that Kim Jong Un also has access to this.
↑ comment by TsviBT · 2025-03-04T07:13:34.960Z · LW(p) · GW(p)
your noble intentions are going to matter only so much once this tech is the public domain
This is somewhat true, yeah. But it's only somewhat true. E.g.
- One can unilaterally make the technology cheaper and more effective. Generally this makes it more widely accessible, which makes it harder for enemies who would want to keep it for themselves to do so. E.g. if inequality were going to be a big resulting problem, you can fix some of it unilaterally in this way.
- Some key aspects of the technology will still require large amounts of skill. I'm thinking in particular of polygenic scores. If KJU wants to make an obedience PGS that actually works for the Korean population, he'd have to find a team of geneticists and psychometricians willing to do so. To say it another way, there is a separate cat to be let out of the bag for each trait (roughly speaking) that you might want to select for/against.
↑ comment by samuelshadrach (xpostah) · 2025-03-04T07:37:49.947Z · LW(p) · GW(p)
I think skill can be stolen via cyberhacking + espionage, assuming you are able to prevent them from just hiring ex employees and ex researchers. The meaningful question for me is how many months of lead time can anyone maintain before they get copied by other nuclear armed countries.
Unless you really find a better plan, my first guess is this is going to lead to an international arms race between multiple countries to develop the most intelligent and politically loyal embryos they possibly can, as fast as they possibly can. The race won’t stop until we hit massively diminishing returns on more investment into both R&D for new genes and raising more children with the genes we already know, and nobody knows how many SDs on which traits we get before this end state.
↑ comment by TsviBT · 2025-03-04T07:49:56.234Z · LW(p) · GW(p)
my first guess is this is going to lead to an international arms race between multiple countries to develop the most intelligent and politically loyal embryos they possibly can, as fast as they possibly can.
I wish there was some more grounded analysis of this sort of thing I could read somewhere. E.g., historical comparisons of other things that states have done with a similar motivation. Or e.g. cases where some technology gets used for good and for evil, and then is it net positive? I feel conversations about what states will do with germline engineering just hit a wall immediately because who knows what will happen.
I think extreme fear of / antipathy towards eugenics is good in part because it constitutes political will to not have states do this sort of thing--controlling people's reproduction, influencing populations. Accordingly, I advocate for genomic emancipation, which is directly opposed to state eugenics.
Replies from: xpostah↑ comment by samuelshadrach (xpostah) · 2025-03-04T07:59:12.494Z · LW(p) · GW(p)
I will let you know when I write an article of this type!
In general though, US policy making circles have a long history of applying just enough pressure on other countries so that the frontier of R&D of every emerging field remains in the US. It’s not a coincidence that frontier of quantum computing and genomics and fusion energy and AI and a hundred other technologies all lie in the US.
Sometimes this does lead to war, US military leaders have afaik started wars over who has nuclear weapons, who has chemical weapons and who has oil. But often there’s more subtle levers that can be pulled such as export controls and backchannel collusion with the leading CEOs of that industry.
I don’t at the moment have a list of examples that doesn’t involve the US, but I know they exist and I agree this is worth writing more on.
↑ comment by TsviBT · 2025-03-04T08:21:05.503Z · LW(p) · GW(p)
From my lay perspective w.r.t. international politics, this seems like it would plausibly be good, to be clear. My frontpage says:
Germline engineering will require an international scientific, technological, and social effort, which we encourage and aim to help with. With that said, as the world's liberal democratic superpower, the United States of America should lead the way on human germline genetic engineering, including enhancement. If America supports this technology while regulating its unsafe and unethical uses, we can show the world how to develop and apply it beneficially.
The US, or at least what the US is supposed to be and isn't impossibly far from, is a place where you could have strong boundaries preventing the government from restricting genomic liberty, while also supporting the development of the tech.
Replies from: xpostah, xpostah↑ comment by samuelshadrach (xpostah) · 2025-03-06T07:09:46.604Z · LW(p) · GW(p)
@TsviBT [LW · GW] I don't know if you were the one who downvoted my comment, but yeah I don't think you've engaged with the strongest version (steelman?) of my critique. Laws (including laws promoting genomic liberty) don't carry the same weight during a cold war as they do during peacetime. Incentives shape culture, culture shapes laws.
And the incentives change significantly when a technology upsets the fundamental balance of power between the world's superpowers.
Replies from: TsviBT, TsviBT↑ comment by TsviBT · 2025-03-06T07:47:58.549Z · LW(p) · GW(p)
Or maybe you're arguing "don't develop any technology" or "don't develop any powerful technology" because "governments might misuse it". That's something you could reasonably argue, but I think you should just argue that in general if that's what you're saying, so the case is clearer.
↑ comment by TsviBT · 2025-03-06T07:35:14.035Z · LW(p) · GW(p)
I didn't downvote any of your comments, and I don't see any upthread comments with any downvotes!
Anyway, you could steelman your case if you like. It might help if you compared to other technologies, like "We should develop powerful thing X but not superficially similar powerful thing Y because X is much worse given that there are governments", or something.
Replies from: xpostah↑ comment by samuelshadrach (xpostah) · 2025-03-06T12:26:40.312Z · LW(p) · GW(p)
Okay!
I'm not universally arguing against all technology. I'm not even saying that an arms race means this tech is not worth pursuing, just be aware you might be starting an arms race.
Intelligence-enhancing technologies (like superintelligent AI, connectome-mapping for whole brain emulation, human genetic engineering for IQ) are worth studying in a separate bracket IMO because a very small differential in intelligence leads to a very large differential in power (offensive and defensive, scientific and business and political, basically every kind of power).
↑ comment by samuelshadrach (xpostah) · 2025-03-04T10:06:02.191Z · LW(p) · GW(p)
Yes it’s possible we end up in a world where the US govt is basically competing with its own shadow yet again. US startup builds some tech, it gets copied 6 months later by non-US startup, US startup feels pressure to move faster as a result and deploys next tech, the next tech too gets copied, etc etc.
I’m not saying this will definitely happen, but there’s a bunch of incentives pushing in this direction.