UML XI: Nearest Neighbor Schemes
post by Rafael Harth (sil-ver) · 2020-02-16T20:30:14.112Z · LW · GW · 3 commentsContents
3 comments
(This is the eleventh post in a sequence on Machine Learning based on this book. Click here [? · GW] for part I.)
Last time, I tried to do something special because the topic was neural networks. [LW · GW] Now we're back to the usual style, but with an unusually easy topic. To fit this theme, it will be a particularly image-heavy post.
The idea of nearest neighbor predictors is to predict the target value of a point based on the target value of the most similar points in the training data. For example, consider a regression problem with and , the following training data, and the new red point:

The position of the points corresponds to their value in . The number next to them corresponds to their target value in . (The four closest points just happen to have target values 1-4 by coincidence.) The other training points also have target values, which are not shown – but since they're real numbers, they are probably things like .
This picture begs the question of how many neighbors to take into account. This is a parameter: in a -nearest neighbor scheme, we consider the target values of the nearest instances. In the instance above, if , we have the following situation:

(Note that the circle just demonstrates which points are closest – we do not choose all points within a fixed distance.)
For , we have two inputs and need to decide what to do with them. For now, we simply declare that the label of our red point will be the mean of all neighbors.

:

:

If the function we're trying to learn here is something like distance from a center and the red point is precisely at that center, then the scheme gets worse and worse the larger becomes. On the other hand, suppose the instance looks like this instead:

where the new point is an unfortunate outlier. In this case, the situation would be somewhat improved for larger . In general, means that every point will get to decide the target values of some new training points (unless there are other training points with identical positions). Here is an image that shows, for each point, the area in which that point will determine the target value of new points given that :

This is also called a Voronoi diagram, and it has some relevance outside of Machine Learning. For example, suppose the red dots are gas stations. Then, for any point, the [red instance determining the cell in which that point lies] represents the closest gas station.
However, for machine learning, this might not be great – just suppose the green point is a crazy outlier. Should it get to decide the target values of new instances in its cell? Another way to describe this problem is that the [function that such a nearest-neighbor predictor will implement] is highly discontinuous.
In general, the more variance there is in the training data, the larger should be, but making larger will, in general, decrease accuracy – the well-known tradeoff. In that sense, one is applying a little bit of prior knowledge to the problem by choosing . But not much – consider the following instance (this time with ):

A linear predictor would probably assign the red point a value that continues the trend – something like . This is because linear models work on the assumption that trends are, in some sense, more likely to continue than to spontaneously revert. Not so with nearest neighbor schemes – if it would get the label 6, and for larger , the label would become smaller rather than larger. In that way, nearest neighbor schemes make weaker assumptions than most other predictors. This fact provides some insight into the question of when they are a good choice for a learning model.
One can also use a weighted average as the prediction, rather than the classical mean. Let's return to our instance, and let's use actual distances:

One way to do this is to weight each prediction proportional to the inverse distance of the respective neighbor. In that case, this would lead to the prediction
where the denominator is there to normalize the term, i.e., make it as if all weights sum up to 1. (You can imagine dividing each weight by the denominator rather than the entire term; then, the weights literally sum up to 1.) In this case, the function we implement would be somewhat more smooth, although each point would still dominate in some small area. To make it properly smooth, one could take the inverse of [the distance plus 1] (thus making the weights range between and rather than and ) and set so that all training points are taken into consideration.
Note that I've been using regression as an example throughout, but decision trees can also be used for classification problems. In that case, each point gets the label which is most popular among its neighbors, as decided by a (weighted) majority vote.
Are there any guarantees we can prove for nearest neighbor schemes?
The question is a bit broad, but as far as sample complexity bounds/error bounds are concerned, the answer is "not without making some assumptions." Continuity of the target function is a necessary condition, but not enough by itself. Consider how much each point tells us about the function we're trying to learn – clearly, it depends on how fast the function changes. Then – suppose our function changes at a pace such that each point provides this amount of information (the green circle denotes the area in which the function has changed "sufficiently little," whatever that means for our case):

In this case, the training data will let us predict new points. But suppose it changes much more quickly:

In this case, we have no chance. In general, for any amount of training data, we can imagine a target function that changes so quickly that we don't have a clue about how it looks on a majority of the area. Thus, in order to derive error bounds, one needs to assume a cap on this rate of change. If you recall the chapter on convexity [LW · GW], this property is precisely -Lipschitzness.
There's a theorem to that effect, but the statement is complicated and the proof is boring. Let's talk about trees. A decision tree is a predictor that looks like this:

(In this case, for a binary classification problem.) If you have never seen a tree before, don't despair – trees are simple. The triangles are called internal nodes, the four endpoints are called leaves, and the lines are called edges. We begin at "Start," evaluate the first "Condition," then move down accordingly – pretty self-explanatory. Trees show up all over the place in Computer Science.
If we allow arbitrary conditions, this is somewhat silly – each predictor can then be represented as a tree via

So the general rule is that the conditions be very simple. That being said, there are several ways one could illustrate that this class is quite expressive. For example, consider a rectangle with labeled sides like this:

The following tree realizes a predictor which labels instances in the rectangle positively and anywhere outside negatively:

The same principle also illustrates how we can implement a logical AND, and OR goes analogously. You may work out how exactly this can be done – however, I don't think those are the best ways to demonstrate what trees are really about. Instead, consider what happens when the tree branches in two:

When the tree branches in two, the domain space is divided in two. And this is true for each branching point, even if the area corresponding to a node isn't the entire domain space to begin with:
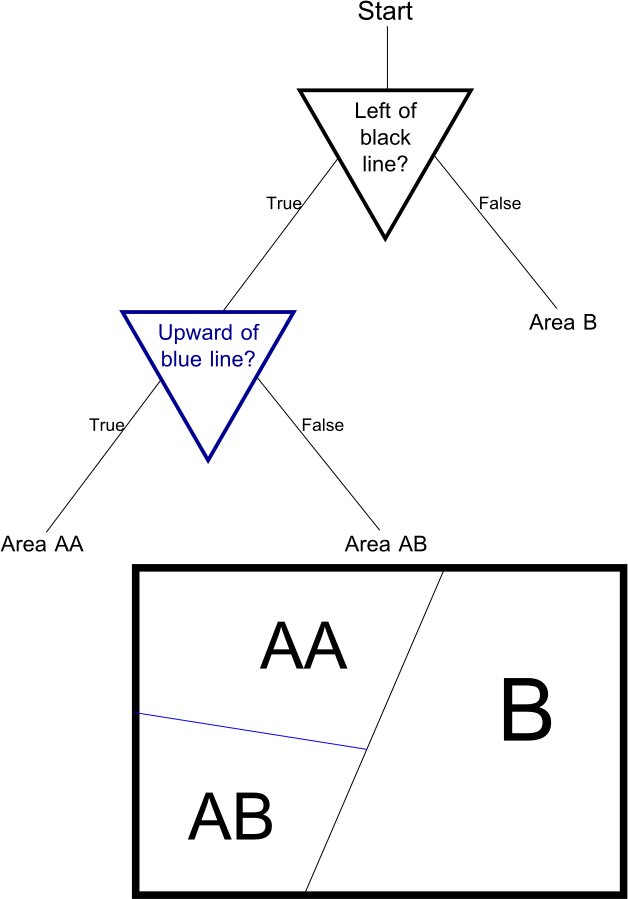
For the initial examples, I have just written down the labels (the 1 and 0 at the leaves) as part of the tree. In reality, they are derived from the training data: for any leaf, the only reasonable label is that which the majority of training points in the corresponding part of the domain space have. And this is why this post isn't called "nearest neighbor and decision trees" – decision trees are a nearest neighbor scheme. The difference lies in how the neighborhoods are constructed: in the classical approach, the neighborhood for each point is based on the distance of every other point to , while in a tree, the neighborhoods are the cells that correspond to the leaves.
In particular, this makes the neighbor-relationship of a tree transitive – if is a neighbor of and is a neighbor of , then is a neighbor of – which is not true for the classical nearest-neighbor approach.
You might recall the concept of VC-dimension from chapter II [LW · GW], which applies for binary classification tasks. To recap: for a given hypothesis class , the VC-dimension is the largest such that there exists a set of domain points which is shattered by . What does it mean for a set to be shattered by ? It means that contains a predictor for any possible labeling combination of the points in . If has elements, these are many combinations. (For example, if , then has to contain a predictor with , a predictor with and also predictors and .) The VC dimension is a measure for the complexity of a class because if all labeling combinations are possible, then learning about the labels of some of the points doesn't tell us the labels of the others. There are both upper- and lower bounds on the sample complexity for classes with finite VC dimension (provided we allow arbitrary probability distributions generating our label points).
This is relevant for decision trees because their VC dimension is trivial to compute...

... it simply equals the number of leaves. This follows from the fact that a tree divides the domain space into subsets where is the number of leaves, as we've just argued. It's equally easy to see that larger subsets cannot be shattered because the tree will assign all points within the same subset the same label.
It follows that the class of arbitrary trees has infinite VC-dimension, whereas the class of trees with depth at most has VC-dimension (the number of leaves doubles with every level we're allowed to go downward).
How do we obtain trees?
Since mathematicians are lazy, we don't like to go out and grow trees ourselves. Instead, we'd like to derive an algorithm that does the hard work for us.
One possibility is a simple greedy algorithm. The term greedy is commonly used in computer science and refers to algorithms that make locally optimal choices. For example, consider the knapsack problem where one is given a number of possible objects that have a weight and a value, and the goal is to pack a subset of them that stays below a particular total weight and has maximum value. A greedy algorithm would start by computing the scores for each item, and start packing the optimal ones.
The general dynamic with greedy algorithms is that there exist cases where they perform poorly, but they tend to perform well in practice. For the knapsack problem, consider the following instance:

A greedy algorithm would start by packing item 3 because it has the best score, at which point the remaining capacity isn't large enough for another item, and the game is over. Meanwhile, packing items 1 & 2 would have been the optimal solution to this problem.
Now we can do something similar for trees. We begin with one of these two trees,

namely, we choose the one that performs better on the training sequence (i.e., if more than half of our training points have label 0, we go with the first tree). After this first step, all remaining steps are the same – we improve the tree by replacing a leaf with a [condition from which two edges go out into two leaves with labels 0 and 1, respectively]. This corresponds to choosing an area of the domain space and dividing it in two.
So let's say we've started with the first tree. We only have one leaf, so the first step will replace that leaf with []:

Now just imagine the same thing starting from an arbitrarily large tree: we take one of its many leaves and substitute the object in the blue circle for it (either exactly the same object or the same except with labels swapped). Somewhere in the domain space, some area that was previously all 0 or all 1 now gets divided into two areas, one with 1 and one with 0.
Since we're running a greedy algorithm, it chooses the split such that the total performance of the tree (on the training data) improves the most – without taking into consideration how this affects future improvements. Of course, we need to restrict ourselves to simple conditions; otherwise, the whole thing becomes pointless – recall this tree:
For example, each restriction could be of the form "bit # of the input point is 1". Many other approaches are possible. Having chosen such a class of restrictions, and also a way to measure how much performance is improved by each restriction, this defines a simple greedy algorithm.
Now, recall that the VC dimension of the class of trees is infinite. Thus, if we let our algorithm run for too long, it will keep growing and growing the tree until it is very large – at that point, it will have overfit the training data significantly, and its true error will probably be quite high. There are several approaches to remedy this problem:
- Terminate the algorithm earlier
- After the algorithm has run to completion, run an algorithm doing the opposite, i.e., cutting down the tree to make it more simple while losing as little performance on the training data as possible
- Grow more trees
To elaborate on the last point: since the problem with an overly large tree is that it learns noise from the training data – i.e., quirks that are only there by chance and don't represent the real world – one way to combat this is by having a bunch of trees and letting them define a predictor by majority vote. This will lead to the noise canceling out, for the same reason that Stochastic Gradient Descent [LW · GW] works. There are, again, at least two ways to do this:
- Use the same tree-growing algorithm, but give it a randomly chosen subset of the training data each time
- Run a modified version of the algorithm several times, and ...
- ... use the all training data each time; but
- ... for each step, choose the optimal splitting point from a randomly chosen subset, rather than from all possible splits
The result is called a random forest.
If we have a random forest, are we still implementing a nearest neighbor scheme? To confirm that the answer is yes, let's work a unified notation for all nearest neighbor schemes.
Suppose we have the training data , and consider a weighting function that says "how much of a neighbor" each training point is to a new domain point. In -nearest neighbor, we will have that iff is one of the nearest neighbors of and 0 otherwise. For weighted -nearest neighbor, if is among the nearest neighbors to , then will be some (in or in , depending on the weighting) determined by how close it is, and otherwise, it will be 0. For a tree, will be 1 iff both and are in the same cell of the partition which the tree has induced on the domain space.
To write the following down in a clean way, let's pretend that we're in a regression problem, i.e., the are values in . You can be assured that it also applies to classification, it's just that it requires to mix in an additional function to realize the majority vote.
Given that we are in a regression problem, we have that
where . Recall that is a learning algorithm in our notation, and is the training data, so is the predictor it outputs.
For a tree, the formula looks the same (but the weighting function is different). And for a forest made out of trees , we'll have different weighting functions , where is the weighting according to tree #. Then for , we have
which can be rewritten as
which can, in turn, be rewritten as
so it is just another nearest-neighbor scheme with weighting function .
3 comments
Comments sorted by top scores.
comment by romeostevensit · 2020-02-17T04:29:28.199Z · LW(p) · GW(p)
I've found it useful to think of quite a lot of algorithms as rearranging the topology of the representation in order to make nodes relevant to a particular decision nearer each other.
Replies from: sil-ver↑ comment by Rafael Harth (sil-ver) · 2020-02-17T08:26:38.480Z · LW(p) · GW(p)
That sounds interesting. Can you share an example other than decision trees?
Replies from: romeostevensit↑ comment by romeostevensit · 2020-02-17T18:20:14.568Z · LW(p) · GW(p)
Anything representable as a graph potentially. Even if I don't wind up using that as the final operation I am doing on it, I find it a useful intermediate step for thinking. E.g. typed edges in a graph, installing a new topology on the graph that changes some of the traversal times.