Project Proposal: Gears of Aging
post by johnswentworth · 2020-05-09T18:47:26.468Z · LW · GW · 43 commentsContents
What Does That Look Like? What’s the Value-Add? Is It Tractable? Outside-View Tractability Summary None 43 comments
Imagine building an airplane the way we do biology research.
Hundreds of separate research groups each pick different airplane parts to work on. There’s no particular coordination in this; people choose based first on what they know how to build, second on what seems fun/interesting/flashy/fashionable, and finally on what they expect to be useful (based on limited local knowledge). Funds are allocated to anything which sounds vaguely related to airplanes. There might be fifty different groups all building propellers, one guy toiling away at gyros and nobody at all on fuel lines. One group is building an autopilot system which could be handy but isn’t really necessary; others are building things which won’t be useful at all but they don’t realize they won’t be useful.
There’s obviously room to generate value by assembling all the parts together, but there’s more to it than that. It’s not just that nobody is assembling the parts, there isn’t even a plan to assemble them. Nobody’s really sure what parts are even being produced, and nobody has a comprehensive list of parts needed in order to build an airplane. If things are missing, nobody knows it. If some work is extraneous, nobody knows that either; nobody knows what the minimum viable path to an airplane looks like. There is no airplane blueprint.
This is what the large majority of aging research looks like. There’s hundreds of different groups each studying specific subsystems. There’s little coordination on what is studied, few-if-any people assembling the parts on a large scale, and nothing like a blueprint.
The eventual vision of the Gears of Aging [? · GW] project is to create a blueprint.
What Does That Look Like?
A blueprint does not have to include all the internals of every single subsystem in comprehensive detail. The idea is to include enough detail that we can calculate whether the airplane will fly under various conditions.
Likewise, a blueprint-analogue for aging should include enough detail that we can calculate whether a given treatment/intervention will cure various age-related diseases, and independently verify each of the model’s assumptions.
Such a calculation doesn’t necessarily involve a lot of numerical precision. If we know the root cause [? · GW] of some age-related disease with high confidence, then we can say that reversing the root cause would cure the disease, without doing much math. On the other hand, we probably do need at least some quantitative precision in order to be highly confident that we’ve identified the root cause, and haven’t missed anything important.
Like an airplane blueprint, the goal is to show how all the components connect - a system-level point of view. Much research has already been published on individual components and their local connections - anything from the elastin -> wrinkles [? · GW] connection to the thymic involution -> T-cell ratio [? · GW] connection to the stress -> sirtuins -> heterochromatin -> genomic instability pathway. A blueprint should summarize the key parameters of each local component and its connections to other components, in a manner suitable for tracing whole chains of cause-and-effect from one end to the other.
Most importantly, a blueprint needs some degree of comprehensiveness. We don’t want to model the entirety of human physiology, but we need a complete end-to-end model of at least some age-related diseases. The more diseases we can fully model, from root cause all the way to observed pathology, the more useful the blueprint will be.
Summary: a blueprint-analogue for aging would walk through every causal link, from root cause to pathology, for one or a few age-related diseases, in enough detail to calculate whether a given intervention would actually cure the disease. See the Gears vs Aging sequence [? · GW] so far for some early pieces working in that direction.
What’s the Value-Add?
Why would a blueprint be useful?
I’d phrase the key feature as “vertical comprehensiveness”, in analogy to vertical integration in economics. It’s mapping out every step of the causal chain from root cause to pathology - the whole “production chain” of one or a few pathologies.
To see why this is useful, let’s compare it to the dual feature: horizontal comprehensiveness. A good example here is the SENS project: a program to prevent aging by cataloguing every potential root cause, and regularly repairing each of them. This is a purely-horizontal approach: it does not require any understanding at all of the causal pathways from root causes to pathologies, but it does require a comprehensive catalogue of every root cause.
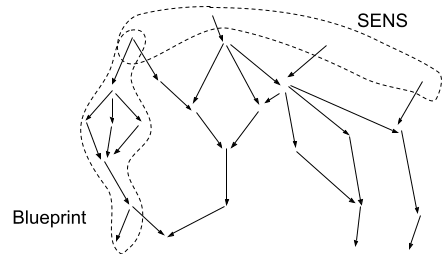
The relative disadvantage of a SENS-style horizontal approach is that there’s no way to check it locally. If it turns out that we missed a root cause, SENS has no built-in way to notice that until the whole project is done and we notice some pathology which hasn’t been fixed. Conversely, if we mistakenly included a root cause which doesn’t matter, we have no built-in way to notice that at all; we waste resources fixing some extraneous problem. For example, here’s the original list of low-level damage types for SENS to address (from the Wikipedia page):
- Accumulation of lysosomal aggregates
- Accumulation of senescent cells
- Age related tumors
- Mitochondrial DNA mutations
- Immune system damage
- Buildup of advanced glycation end-products
- Accumulation of extracellular protein aggregates
- Cell loss
- Hormonal muscle damage
Note the inclusion of senescent cells. Today, it is clear that senescent cells are not a root cause of aging, since they turn over on a timescale of days to weeks. Senescent cells are an extraneous target. Furthermore, since senescent cell counts do increase with age, there must also be some root cause upstream of that increase - and it seems unlikely to be any of the other items on the original SENS list. Some root cause is missing. If we attempted to address aging by removing senescent cells (via senolytics), whatever root cause induces the increase in senescent cells in the first place would presumably continue to accumulate, requiring ever-larger doses of senolytics until the senolytic dosage itself approached toxicity - along with whatever other problems the root cause induced.
This isn’t to bash the SENS program; I’m personally a fan of it. The point is that the SENS program lacks a built-in way to cheaply verify its plan. It needs to rely on other kinds of research in order to make sure that its list of targets is complete and minimal.
Conversely, built-in verification is exactly where vertical comprehensiveness shines.
When we have a full causal pathway, we can ask at each step:
- Does this causal relationship actually hold?
- Do the immediate causes actually change with age by the right amount to explain the observed effects?
Because we can do this locally, at each step of the chain, we can verify our model as we go. Much like a mathematical proof, we can check each step of our model along the way; we don’t need to finish the entire project in order to check our work.
In particular, this gives us a natural mechanism to notice missing or extraneous pieces. Checking whether senescent cells are actually a root cause is automatically part of the approach. So is figuring out what’s upstream of their age-related increase in count. If there’s more than one factor upstream of some pathology, we can automatically detect any we missed by quantitatively checking whether the observed change in causes accounts for the observed change in effects.
Summary: the main value-add of a blueprint-style end-to-end model is that we can locally verify each link in the causal chain, usually using already-existing data.
Is It Tractable?
I think that the data required to figure out the gears of most major human age-related diseases is probably already available, online, today. And I don’t mean that in the sense of “a superintelligent AI could figure it out”; I mean that humans could probably figure it out without any more data than we currently have.
That belief stems mainly from having dug into the problem a fair bit already. Everywhere I look, there’s plenty of data. Someone has experimentally tested, if not the exact thing I want to know, at least something close enough to provide evidence.
The hard part is not lack of data, the hard part is too much data. There’s more than a human could ever hope to work through, for each major subsystem. It’s all about figuring out which questions to ask, guessing which experiments could provide evidence for those questions and are likely to have already been done, then tracking down the results from those experiments.
So I think the data is there.
The other piece of tractability is whether the system is simple enough, on some level, that a human can hope to understand all the key pieces. Based on having seen a fair bit, I definitely expect that it is simple enough - not simple, there are a lot of moving pieces and figuring them all out takes a fair bit of work, but still well within human capacity. We could also make some outside view arguments supporting this view - for instance, since the vast majority of molecules/structures/cells in a human turn over on much faster timescales than aging, there are unlikely to be more than a handful of independent root causes.
Outside-View Tractability
If a project like this is both useful and tractable, why hasn’t it already been done?
The usual academic outlet for a blueprint-style vertically-comprehensive work would be a textbook. And there are textbooks on aging, as well as monographs, and of course books on various subtopics as well. Unfortunately, the field is still relatively young, and textbook-writing tends to be under-incentivized in the sciences; most academic hiring and tenure committees prefer original research. Even those textbooks which do exist tend to either involve a broad-but-shallow summary of existing research (for single-author books) or standalone essays on particular components (for multi-author monographs). They are part catalogues, not blueprints.
But the biggest shortcoming of typical textbooks, compared to the blueprint-style picture, is that typical textbooks do not actually perform the local verification of model components.
This is exactly the sort of problem where we’d expect a rationalist skillset - statistics, causality, noticing confusion, mysterious answers, etc - to be more of a limiting factor than biological know-how. Add that to the lack of incentive for this sort of work, and it’s not surprising that it hasn’t been done.
A handful of examples, to illustrate the sort of reasoning which is lacking in most books on aging:
- Many review articles and textbooks claim that the increased stiffness of blood vessels in old age results (at least partially) from an increase in the amount of collagen relative to elastin in vessel walls. But if we go look for studies which directly measure the collagen:elastin ratio in the blood vessels, we mostly find no significant change with age (rat, human, rat).
- Many reviews and textbooks mention that the bulk of reactive oxygen species (ROS) are produced by mitochondria. Attempts at direct measurement instead suggest that mitochondria account for about 15% (PhysAging, table 5.3).
- In 1991, a small-count genetic study suggested that amyloid protein aggregates in the brain cause Alzheimers. Notably, they “confirmed diagnoses via autopsy” - which usually means checking the brain for amyloid deposits. At least as early as 2003, it was known that amyloid deposits turn over on a timescale of hours. Yet, according to Wikipedia, over 200 clinical trials attempted to cure Alzheimers by clearing plaques between 2002 and 2012; only a single trial ended in FDA approval, and we still don’t have a full cure.
- A great deal of effort has gone into imaging neuromuscular junctions in aging organisms. As far as I can tell, there was never any significant evidence that the observed changes played any significant causal role in any age-related disease. They did produce really cool pictures, though.
These are the sorts of things which jump out when we ask, for every link in a hypothesized causal chain:
- Does this causal relationship actually hold?
- Do the immediate causes actually change with age by the right amount to explain the observed effects?
Summary
I think that the data required to figure out the gears of most major human age-related diseases is probably already available, online, today. The parts to build an airplane are already on the market. We lack a blueprint: an end-to-end model of age-related pathologies, containing enough detail for each causal link in the chain to be independently validated by experimental and observational data, and sufficient to calculate whether a given intervention will actually cure the disease.
43 comments
Comments sorted by top scores.
comment by Shmi (shminux) · 2020-05-09T22:45:57.027Z · LW(p) · GW(p)
I think that the data required to figure out the gears of most major human age-related diseases is probably already available, online, today.
I find that wildly optimistic. My guess is quite opposite: while we see the symptoms of aging, and have some inkling of the underlying processes, like telomerase shortening, we have no clue at all about the underlying reasons for it, let alone about how this can be changed in a way that prolongs youth and delays the onset of aging.
Replies from: Natália Mendonça↑ comment by Natália (Natália Mendonça) · 2020-05-11T01:45:01.156Z · LW(p) · GW(p)
I don’t see how that contradicts his claim. Having the data required to figure out X is really not the same as knowing X.
comment by emanuele ascani (emanuele-ascani) · 2020-05-10T10:46:07.906Z · LW(p) · GW(p)
I really like this post. I think it is probably also relevant from an Effective Altruism standpoint (you identify a tractable and neglected approach which might have a big impact). I think you should probably crosspost this on the EA Forum, and think about if your other articles on the topic are apt to be published there. What do you think?
If you read my profile both here and on the EA Forum you'll find a lot of articles in which I'm trying to evaluate aging research. I'm making this suggestion because I think you are adding useful pieces.
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-10T15:50:09.547Z · LW(p) · GW(p)
Thanks, this is a great suggestion I hadn't thought of. I'll probably do it.
comment by Vaniver · 2020-05-11T03:17:45.866Z · LW(p) · GW(p)
Agreed on the general point that having an overall blueprint is sensible, and that any particular list of targets implies an underlying model.
Note the inclusion of senescent cells. Today, it is clear that senescent cells are not a root cause of aging, since they turn over on a timescale of days to weeks. Senescent cells are an extraneous target. Furthermore, since senescent cell counts do increase with age, there must also be some root cause upstream of that increase - and it seems unlikely to be any of the other items on the original SENS list. Some root cause is missing. If we attempted to address aging by removing senescent cells (via senolytics), whatever root cause induces the increase in senescent cells in the first place would presumably continue to accumulate, requiring ever-larger doses of senolytics until the senolytic dosage itself approached toxicity - along with whatever other problems the root cause induced.
I think this paper ends up supporting this conclusion, but the reasoning as summarized here is wrong. That they turn over on a timescale of days to weeks is immaterial; the core reason to be suspicious of senolytics as actual cure is that this paper finds that the production rate is linearly increasing with age and the removal rate doesn't keep up. (In their best-fit model, the removal rate just depends on the fraction of senolytic cells.) Under that model, if you take senolytics and clear out all of your senescent cells, the removal rate bounces back, but the production rate is steadily increasing.
You wouldn't have this result for different models--if, for example, the production rate didn't depend on age and the removal rate did. You would still see senescent cells turning over on a timescale of days to weeks, but you would be able to use senolytics to replace the natural removal process, and that would be sustainable at steady state.
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-11T03:41:18.552Z · LW(p) · GW(p)
Great point, and nice catch.
comment by DirectedEvolution (AllAmericanBreakfast) · 2022-10-22T07:25:11.232Z · LW(p) · GW(p)
My simple model of aging is a shifting balance between bounded, specialized repair mechanisms and unbounded, combinatorial forms of damage.
Organisms begin life as single cells. They undergo selection for a viably low level of starting damage beginning with mitosis or meiosis.
At all times, the environment and the byproducts of normal intracellular processes cause damage to cells and their surroundings. Many of these factors cause predictable forms of damage. UV light, for example, causes two specific types of DNA lesions, and cells have specialized DNA repair mechanisms to fix them. If all damage to cells and their environments was of a type and amount that was within repair capacities of organisms, then aging would not occur.
Repair is limited to specific, evolved capacities maintained by organisms to deal with the most fitness-reducing forms of damage they face. Outside these narrow capacities, repair mechanisms are typically useless. In contrast, damage factors have access to the entire energy landscape, and so they have access to an unbounded and combinatorial set of damage vectors that far outstrips the total repair capacity of the organism. We can break this down into two components.
- Unusual damage: Common individual factors with mostly predictable effects can sometimes inflict unusual forms of damage, for which are uncommon enough that it is not evolutionarily tractable for organisms to evolve and maintain a repair infrastructure. Uncommong individual damage factors can likewise inflict forms of damage for which no repair mechanism exists.
- Damage systems: Individual damage factors can have emergent damaging effects in combination, and the number of potential combinations means that this becomes another source of damage beyond the bounds of existing repair infrastructure. Forms of damage may accumulate at a faster rate than they can be repaired if a system of damage factors effects a positive feedback loop, inhibits repair mechanisms, or evades them.
How does life survive in this context, where damage factors always have the advantage over repair mechanisms? First of all, it frequently doesn't. Nonviable eggs and sperm or unicellular organisms die silently. The biological machinery of reproduction, including the repair and protection mechanisms that shelter it, do not always successfully survive to the time of replication, and if they do not, that line dies out.
When life does survive to maturity, it by definition has been selected for viability - successful shelter by normal prevention and repair mechanisms from excessive unusual or combinatorial damage.
What does this model predict?
- An "irreparable damage landscape" will exist beyond the boundaries of what a species' repair mechanisms can fix; these repair mechanisms are dictated by evolutionary constraints. There will be common and uncommon forms of irreparable damage. Biomedicine can supplement normal repair mechanisms with artificial ones. There will be much that science and engineering can accomplish to push the "repair boundary" further out than what evolution alone permits.
- Some specific forms of irreparable damage will be central nodes on a damage DAG, giving rise to diverse damaging downstream effects that may be the direct causes of morbidity and mortality. Tackling these central nodes will result in a higher payoff in terms of healthspan and lifespan than tackling the downstream effects. However, there is no single, fundamental cause of the damage associated with aging.
- Whether or not longevity interventions are tractable will shed light on the optimizing efficiency of evolution. If evolution is efficient, then longevity interventions will come with steep tradeoffs, require complex feats of engineering, or be forced to address a multitude of minor forms of irreparable damage for which it is not worthwhile for evolution to maintain repair mechanisms. If evolution is inefficient, then we will find many significant damage nodes beyond the repair boundary that have simple solutions, yield major gains in lifespan and healthspan, and prevent or mitigate many diseases.
- Longevity research is not fundamentally different from research into known diseases. Cancer, diabetes, and heart disease all have early, late, and pre-detectable stages. Longevity research is really an investigation into the pre-detectable stage of these diseases. When we try to treat late-stage cancer, we are far from longevity research. When we try to understand what activates dormant prostate cancer cells in the bone marrow, we are closer to longevity research. When we study what causes those prostate cancer cells to home to the bone marrow and how they survive there, we are closer still. When we examine what chemical reactions tend to give rise to the genetic mutations leading to prostate cancer, we are even more centrally doing longevity research. There is a temptation to assume that if we trace back the causal pathway far enough, we will find a monocause. This is not so. Many causes may input into a specific node, and a specific node may point out at many other nodes. The arrow of causality can point in two directions as well, or form loops between several nodes.
- The bottleneck for longevity research will be the ability to show predictive validity of early damage factors leading to later symptoms. For example, being able to detect a cluster of genetic or chemical changes that will lead to prostate cancer with 80% confidence 5 years before it occurs. This is challenging both because increasingly early predictions have more nodes in the causal link, likely require more data to achieve a given level of predictive validity, require more interventions to correct the problem, and also because the earlier we make the prediction, the slower the feedback loop. Importantly, however, these challenges are relative. Early prediction will be harder than later prediction, but it does not mean that it will be hard. This depends on the efficiency of evolution. If it is a good optimizer for longevity, then the problem of early prediction will be hard. If it is a poor optimizer, we can hope to find some easy wins.
- Diversity, noise, and ambiguity in biological systems and our methods of measurement will be key problems in longevity research.
- Even if evolution is a good optimizer, we have a much greater ability to control our environments than we did in the recent past. If evolution has optimized our bodies for robustness to environmental stressors that are no longer a serious concern by making tradeoffs against longevity, then we can get alpha by re-engineering our bodies in ways that sacrifice this unnecessary robustness in exchange for optimizing for the most longevity-promoting environments we can create.
- Again, longevity research in many cases will look like a specific subtype of normal disease research. My own PI and his wife, among others, examine the pre-metastatic niche. How does a local site become adapted to colonization by metastatic cancer cells? Trace this back further. What initiates that process of adaptation?
- Some damage nodes will have so many common and equally important causes that it is not tractable to trace back to their causes and address them. Yet it could be worthwhile if that node also has many downstream effects.
- Some biological events may cause damage in some contexts and be necessary for life in others, or both at the same time. The objective is always to repair or prevent damage with an acceptable burden of side effect damage.
- All this suggests a simple strategy for doing effective longevity research: trace back diseases to their earliest causes, and see if the cause of a particular disease might be a branch point to multiple other diseases. If you have identified such a branch point, attack it.
↑ comment by DirectedEvolution (AllAmericanBreakfast) · 2022-10-24T00:50:30.636Z · LW(p) · GW(p)
Following up based on John's points about turnover, we can add in a "template decay" aspect to this model. The body has many templates: DNA, of course, but also stem cells, and tissue architectures, possibly among others. Templates offer a highly but not perfectly durable repository of information or, more intuitively in some cases, structural guidance for turnover and regeneration processes.
When damage factors impact the downstream products of various templates (mRNA and proteins, differentiated cells, organs), turnover initiated by the upstream template can mitigate or eliminate the downstream impact. For example, protein damage can be repaired by degredation in proteasomes, differentiated cell dysfunction can be repaired by apoptosis, stem cell proliferation and differentiation into a replacement for the dysfunctional cell. Wounds can be regenerated as long as sufficient local tissue architecture remains intact to guide tissue reconstruction.
But templates cannot themselves be repaired unless there is a higher-order upstream template from which to initiate this repair. A key question is redundancy. DNA has no redundancy within the cell, but stem cells could potentially offer that redundancy at the level of the cell. If one stem cell dies or becomes senescent, can another stem cell copy itself and replace it? Yet we know stem cells age and lose proliferative and differentiation capacity. And this redundancy would only provide a lasting solution if there was a selecting force to regulate compatibility with host architecture instead of turning into cancer. So whatever redundancy they offer, it's not enough for long-term stem cell health.
For an individual's DNA sequence at least, durable storage is not a bottleneck. We are challenged to provide durable external repositories for other forms of information, such as tissue architecture and epigenetic information. We also lack adequate capacity to use any such stored information to perform "engineered turnover."
We might categorize medicine in two categories under this paradigm:
- Delay of deterioration, which would encompass much of the entire current medical paradigm. Chemo delays deterioration due to cancer. Vaccines and antibiotics prevent deterioration due to infection. Seatbelts prevent acute deterioration due to injury from car crashes. Low-dose rapamycin may be a general preventative of deterioration, as is exercise and good diet.
- Template restoration, which has a few examples in current medicine, such as organ, tissue, and fecal transplants. Gene therapy is a second example. Some surgeries that reposition tissues to facilitate a new equilibrium of well-formed growth is a third example. If it becomes possible to de-age stem cells, or to replace DNA that has mutated with age with the youthful template, these interventions would also be categorizwed as template restoration.
- Template replacement. An example is an artificial heart, which is based on a fundamentally different template than a biological heart, and is subject to entirely different deterioration dynamics and restoration possibilities.
The ultimate aim is to apply organized energy from outside the patient's system, primarily in the form of biomedical interventions, to create a more stringent selection force within the patient's body for a molecular, cellular, and tissue architecture compatible with long-term health and survival of the patient. In theory, it ought to be possible to make this selection force so stringent that there is no hard limit to the patient's lifespan.
By this point, "there's no such thing as a cure for cancer" is a cliche. But I don't think this is necessarily true. If we can increase the stringency of selection for normal, healthy cells and against the formation of cancer, then we can effectively eliminate it. Our current medicine does not have the ability to target asymptomatic accumulated cellular disorder, the generator of cancer, for repair or replacement. When we can do this, we will have effectively cured cancer, along with a host of other diseases.
De-aging tissue can be conceived of as:
- Delivering an intervention to host cells in situ to effect repair, such as a gene therapy that identifies and replaces mutated DNA in host cells with the original sequence.
- Extracting healthy cells from the host, proliferating them in vitro, and transplanting them back into the host, either as cells or in the form of engineered tissues.
- Replacing tissue with synthetic or cybernetic constructs that accomplish the same function, such as hip replacements, the destination artificial heart, and (someday soon) the bioartificial kidney.
None of this will be easy. Implanted stem cells and gene therapy can both trigger cancer at this stage in our technological development. Our bioproduction capacities are limited - many protocols are limited by our ability to culture a sufficient quantity of cells. But these are all tractable problems with short feedback loops.
As John points out, the key problem is that too many of our resources are not being applied to the right bottlenecks, and there isn't quite enough of a cohesive blueprint or plan for how all these interventions will come together and result in longevity escape velocity. But at the same time, I tend to be pretty impressed with the research strategy of the lab directors I've spoken with.
↑ comment by johnswentworth · 2022-10-22T21:38:20.962Z · LW(p) · GW(p)
I think the main key concept missing here is that turnover "fixes" basically any damage in turned-over components by default, even when there's not a specific mechanism for that damage type. So, protein turnover "fixes" basically any damage to a protein by replacing the whole protein, cell turnover "fixes basically any damage to a cell by replacing the whole cell, etc. And since the vast majority of most multicellular organisms turns over regularly, at multiple scales (e.g. even long-lived cells have most of their individual parts turn over regularly), that means the vast majority of damage gets "fixed" even without a specific mechanism.
The key question is then: which things don't turn over quickly, and what kind of damage do they accumulate?
Replies from: AllAmericanBreakfast↑ comment by DirectedEvolution (AllAmericanBreakfast) · 2022-10-22T22:56:46.702Z · LW(p) · GW(p)
It seems like a key question is something like the rate of turnover vs. the rate of damage proliferation. We also need to factor in the potential for damage to inhibit turnover (among other repair mechanisms). After all, turnover is a complex set of processes involving identification, transport, destruction, and elimination of specific structures. The mechanisms by which it is effected are subject to damage and disrepair.
We also need to consider the accuracy of regeneration after turnover. When a cell dies and is replaced, the new cell will not anchor in the exact same position in the ECM. On larger scales, this might average out, but it might also lead to larger anatomical dysfunction. Individuals that survive from meiosis into maturity may do so because they've been lucky in avoiding random anatomical fluctuations that kill their less fortunate brethren. If the body does not have adequate mechanisms to maintain precise numbers, arrangements, and structures of organelles, ECM, tissue, and gross anatomy over time, then turnover won't be able to solve this problem of "structural decay."
This is to some extent true of the proteins and cells of the body. Malformed cells and proteins are targeted for destruction. So the survivors are similarly selected for compatibility with the body's damage detection mechanisms, even though the best way of being compatible with those mechanisms is to evade them entirely.
So we have several things to investigate:
- Structures with a slower rate of turnover than of damage accumulation
- Example: cortical neurons, lens proteins
- Forms of damage that impair or evade turnover
- Example: mutations/epigenetic changes that eliminate apoptosis receptors in cancer cells, inert protein aggregates that cannot be degraded by the proteasome and accumulate in the cell, preventing turnover of yet other proteins. As a second example of protein evasion of turnover, loss of lysine or methionine would eliminate a site for ubiquitination, impairing the marking of proteins for destruction by the proteasome.
- Forms of damage that accelerate damage proliferation
- Example: metastatic cancer
- Forms of damage that turnover cannot fix, such as structural decay.
- Example: perhaps thymic involution? I am not sure, but I am confident that this is a real phenomenon.
That said, I think it's a very valuable insight to keep in mind that for any form of damage, we always have to ask, "why can't turnover fix this problem?"
comment by Viliam · 2020-05-10T18:05:37.770Z · LW(p) · GW(p)
Thanks for working on an important problem!
I am afraid that the true answer will turn out to be: There are thousands of mechanisms that gradually kill you after hundred years or so. They are side effects of something else, or maybe trade-offs like "this will make your muscles 0.01% stronger or faster, but they will accumulate some irrepairable damage that will ruin them in hundred years". They are not selected against by natural selection, because for each of them individually, the chance that this specific mechanism will be the only cause of your death is very small.
At least it seems to me that if a new mutation would arise, that would provide you some small advantage, in return for certainly killing you when you are 150, the evolution would happily spread it. And this probably kept happening since the first multicellular organisms have appeared. EDIT: Okay, probably not.
Replies from: johnswentworth, None↑ comment by johnswentworth · 2020-05-11T00:44:14.700Z · LW(p) · GW(p)
There are some reasons for optimism, although nothing that conclusively rules out a large number of mechanisms.
First, humans only have ~tens of thousands of genes, so thousands of independent mechanisms would mean that a substantial portion of all the molecular species in the organism are involved. On top of that, the vast majority of components - from molecules to organelles to cells - turn over on timescales many orders of magnitude faster than aging, which severely limits the number of possible root mechanisms. Just based on that, I'd be very surprised to see even hundreds of independent mechanisms. Tens of mechanisms would be plausible, but single-digits seems most likely. It's just extremely rare for biological systems to operate on timescales that slow.
Another direction: there are animals which display negligible senescence (i.e. no aging). Most of them are not very close to humans on the evolutionary tree, but even distant animals have reasonably similar core mechanics, so there's only so much room for divergence.
Replies from: Roko↑ comment by Roko · 2020-05-22T16:39:08.663Z · LW(p) · GW(p)
which animals?
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-22T17:04:17.530Z · LW(p) · GW(p)
See Highlights of Comparative and Evolutionary Aging [? · GW].
↑ comment by [deleted] · 2020-05-10T18:36:52.040Z · LW(p) · GW(p)
(Not the author, obviously.) Part of my personal intuition against this view is that even amongst mammals, lifespans and the way in which lives ends seems to vary quite a bit. See, for example, the biological immortality Wikipedia page, this article about sea sponges and bowhead whales, and this one about naked mole rats.
That said, it's still possible we're locked in a very tricky-to-get-out-of local optima in a high dimensional space that makes it very hard for us to make local improvements. But then I suspect OP's response would be that the way to get out of local optima is to understand gears.
Replies from: Viliam↑ comment by Viliam · 2020-05-11T14:20:52.274Z · LW(p) · GW(p)
I agree with both replies, that if there are species, including mammals, which live very long, then the mechanisms responsible for killing us have not been here "since the first multicellular organisms have appeared".
Was aging death reinvented independently by so many species, because statistically, at some moment most of them encountered a mutation that provided a Faustian deal of an advantage X, in return for death at age N (for various values of X and N)? And the few immortal ones are simply those who avoided this class of mutations (i.e. had the luck that any mutation causing death turned out to be a net disadvantage)?
Or are the mechanisms of aging death more general, and there are a few species which found out how to avoid them? In other words, are the immortal species descendants of mortal species? Is there a chance to find the answer in archaelogical record? (Whether the common ancestor of a mortal species and an immortal species was mortal or immortal itself.) I'd like to see the entire evolutionary tree, with colors, like these species are/were mortal, these were not.
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-11T16:24:00.947Z · LW(p) · GW(p)
Or are the mechanisms of aging death more general, and there are a few species which found out how to avoid them?
There's a fun question. One of the main things which spurred the current wave of aging research was the discovery that certain interventions increase lifespan across a huge range of different species (e.g. calorie restricted diets). That strongly suggests conserved mechanisms, although it doesn't rule out some species-specific mechanisms also operating in parallel.
I haven't looked this up, but I'm fairly confident the negligibly-senescent species are descendents of species which age. Examples of negligibly-senescent species include some turtles, rougheye rockfish, and naked mole rats; I'm pretty sure the closest relatives of most of those species do age. Again, I don't have numbers, but I have the general impression that negligible senescence is an unusual trait which occurs in a handful of isolated species scattered around the evolutionary tree.
comment by Michael Edward Johnson (michael-edward-johnson) · 2020-05-10T05:33:21.594Z · LW(p) · GW(p)
I think there's a lot of great points and habits of thinking here. My intuition is that it's really hard to marshall resources in this space, SENS has done this, and they seem pretty open to feedback (I think Jim is great). The next exploratory step in getting this to happen might be as a proposal to SENS.
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-10T15:50:50.147Z · LW(p) · GW(p)
Great suggestion, I'll likely look into it.
Replies from: gworley↑ comment by Gordon Seidoh Worley (gworley) · 2020-05-13T03:22:56.730Z · LW(p) · GW(p)
I was going to come here to say to look at SENS. I feel like they do have an idea of a "blueprint" for solving aging and are working on executing on it, and they are from my outside perspective working in a way similar to MIRI: they have a problem to solve, some ideas about how to do it, and some research agendas they are executing on to move them towards the answer.
Radiation out from SENS is a constellation of folks working on this stuff. I'm not sure, though, that these folks also lack a blueprint, only that they work in field where they can't easily publicly claim to have one.
comment by DirectedEvolution (AllAmericanBreakfast) · 2022-10-23T03:29:08.874Z · LW(p) · GW(p)
Another relevant paper is Effect of aging on stem cells. Stem cells are the foundation of cell turnover. Yet their capacity to proliferate and differentiate deteriorates with age. This seems like it's getting closer to a root cause of breakdown of a major aging defense mechanism. Might be a high-value node to understand and prevent aging.
Edit: I see you covered this to some extent in your "Core Pathways of Aging" post.
comment by DirectedEvolution (AllAmericanBreakfast) · 2022-10-23T01:21:15.106Z · LW(p) · GW(p)
In case you haven't seen it, a 2007 model suggests that turnover rates have a complex relationship with cancer and aging. Mutations during cell division would tend to make high turnover rates cause both aging and cancer. Mutations that are caused by DNA damage independently of cell division tend to make high turnover rates protective against aging, but cancer-promoting. I've only looked at the abstract so far. If that aspect of the turnover/mutation relationship holds and turns out to be the most important way turnover relates to aging, then it becomes hard to say intuitively whether cell turnover is on net protective or promoting of aging.
Some questions I have before a deeper dive into the paper:
- Does the model address the function of turnover in selecting against internally disorganized cells?
- Does the model address mechanisms that select against dysfunctionally mutated cells?
- Does the model address disorder introduced by imprecise replacement of the old cells, either in terms of the organization or the location of the replacement cell?
comment by ChristianKl · 2020-05-11T08:58:25.433Z · LW(p) · GW(p)
At least as early as 2003, it was known that amyloid deposits turn over on a timescale of hours. Yet, according to Wikipedia, over 200 clinical trials attempted to cure Alzheimers by clearing plaques between 2002 and 2012;
To me that study seems to say that the half-life of the amyloid protein was higher in those those mice with amyloid deposists.
That's compatible with the thesis that alzheimers patients have a problem with clearing the amyloid protein fast enough and that a drug that increases their ability to clear the protein would be helpful.
The comparison between airplane engineering and biology research doesn't seem fitting to me. While academic research happens in an unstructured way, we do have various big pharma companies who's organisation is closer to how an airplane company does engineering.
We also have plenty of academic physics research that provided us better insight into how to build good planes.
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-11T16:13:27.396Z · LW(p) · GW(p)
That's compatible with the thesis that alzheimers patients have a problem with clearing the amyloid protein fast enough and that a drug that increases their ability to clear the protein would be helpful.
Correct. But the turnover time does still conclusively rule out amyloid plaques as a root cause, which to my understanding was the main hypothesis which made those studies look like a good idea in the first place. If the plaques aren't a root cause, then the next question is whether they mediate the effect, and I don't think the genetics study provided any evidence at all relevant to that question (since they didn't even separate the measurement of plaque from dementia). This is all based on a cursory reading of that research, though, so take it with a grain of salt.
As for the analogy between big pharma companies and airplane companies... a better analogy might be to a company which attempts to make airplanes by searching for objects which look light and floaty, throwing them into the air to see if they fly, p-hacking as much as they can get away with to convince regulators that they can fly, and then marketing them as airplanes.
Replies from: ChristianKl↑ comment by ChristianKl · 2020-05-12T11:30:05.043Z · LW(p) · GW(p)
I don't think that believing in root causes motivate such interventions. Complex systems with feedback loops don't necessarily have root causes.
To the extend that big pharma throws objects in the air to see whether they fly, it's because they millions they spent to evaluate whether the object is a good candidate are not enough to analyse the existing literature to get to root causes.
In addition to big pharma we have billions going into Calico to reason in a structured way about where to attack aging.
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-12T14:46:52.976Z · LW(p) · GW(p)
A system with feedback loops generally equilibrates on roughly the timescale of its slowest component, so in that case we're using "root cause" to refer to that slowest component. See Homeostasis and "Root Causes" in Aging [? · GW].
... millions they spent to evaluate whether the object is a good candidate are not enough to analyse the existing literature to get to root causes.
I don't think that big pharma actually makes all that strong an effort to understand root causes, nor are they particularly incentivized to do so. The way their market is structured, even a marginally-effective drug can be sold at a high price, with lots of marketing, and lots of people will use it because there just aren't good options and people want something. It's the signalling theory of medicine. Even to the extent that pharma companies are incentivized to hunt for root causes, it's the sort of problem which is inherently hard to hire for [LW · GW] - someone who doesn't already understand how to structure such investigations won't be able to distinguish actual competence from a top-tier PhD. Much easier to invest in p-hacking plus marketing.
(They do have an incentive to look like they invest in understanding root causes, though, which is a great use for those top-tier PhDs. The executives probably even believe it.)
As to Calico, my understanding is that they mostly throw black-box ML at large biological datasets. That's not a very good way to work out the gears of biological systems [LW · GW], although it is very shiny.
Replies from: ChristianKl↑ comment by ChristianKl · 2020-05-13T07:50:11.251Z · LW(p) · GW(p)
A system with feedback loops generally equilibrates on roughly the timescale of its slowest component
When it comes to illnesses, the system frequently is not in an equilibrium. Alzheimers in particular gets worse over time till the person is dead.
I don't think that big pharma actually makes all that strong an effort to understand root causes, nor are they particularly incentivized to do so
Clinical trials are very expensive. To the extend that you can spend a few million to choose promising candidates for your clinical trials, big pharma is incentivized to spend that money.
FDA approvals aren't as easily P-hacked given that you need multiple studies that come to the same result as academic papers.
It might be true that currently neither big pharma nor Calico employs people who are actually well qualified to run the exploration, but that's similar to employing someone who's bad at airplane design to design your new airplane.
Replies from: PeterMcCluskey, johnswentworth↑ comment by PeterMcCluskey · 2020-05-14T16:52:47.900Z · LW(p) · GW(p)
People who fund billion dollar trials normally demand that they be run by relatively cautious people. That conflicts with the need for innovative models.
The good news is that there's wide variation in how expensive trials need to be. The larger the effect size, the smaller the trial can be - see the TRIIM trial.
↑ comment by johnswentworth · 2020-05-13T14:42:09.362Z · LW(p) · GW(p)
When it comes to illnesses, the system frequently is not in an equilibrium. Alzheimers in particular gets worse over time till the person is dead.
Yes, that would be an equilibration timescale of infinity. The "slowest component" never reaches equilibrium at all.
comment by Bucky · 2020-05-10T19:26:35.855Z · LW(p) · GW(p)
What are your estimates for how many nodes / causal relationships you would need to investigate to figure out one blueprint?
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-10T21:57:35.122Z · LW(p) · GW(p)
The very simplest would be something like the lens [? · GW], where there's a single connection from an obvious root cause to a pathology.
But the real question is how many steps are involved in the "core" diseases of aging, i.e. the stuff that all shows up in progerias. I would guess on the order of ~10 steps would be the maximum depth, but there's a lot of fan-out; any particular disease would involve another ~10 key pieces as an upper estimate . That includes things which don't change with age but need to be understood as experimental knobs; it does not include red herrings.
comment by Alex K. Chen (parrot) (alex-k-chen) · 2020-05-14T04:00:55.327Z · LW(p) · GW(p)
Like an airplane blueprint, the goal is to show how all the components connect - a system-level point of view. Much research has already been published on individual components and their local connections - anything from the elastin -> wrinkles [? · GW] connection to the thymic involution -> T-cell ratio [? · GW] connection to the stress -> sirtuins -> heterochromatin -> genomic instability pathway. A blueprint should summarize the key parameters of each local component and its connections to other components, in a manner suitable for tracing whole chains of cause-and-effect from one end to the other.
aren't there better methods of characterizing these connecting components than a textbook? Textbooks are super-linear and ill-suited for complex demands where you want to do things like "cite/search for all examples of H3K27 trimethylation affecting aging in each and every studied model organism". They're not great for characterizing all the numerous rare-gene variants and SNPs that may help (such as, say, the SNP substitution in bowhead whale and kakapo p53 and how this single SNP *mechanistically* affects interactions between p53 and all the other downstream effects of p53 - such as whether it increases/decreases/etc). There are many databases of aging already (esp those compiled by JP de Magalhaes and the ones recently outputted by the Glen-Corey lab and Nathan Batisty senescent cell database) but the giant databases return giant lists of genes/associations and effect sizes but also contain no insight in them.
The aging field moves fast and there are already zillions of previous textbooks that people don't read anymore simply b/c they expect a lot of redundancy on top of what they already know.
In particular I'd like a database that lists prominent anomalies/observations (eg naked mole rat enhanced proteasome function or naked mole rat enhanced translational fidelity or "naked mole rat extreme cancer resistance which is removed if you ablate [certain gene]") which then could be made searchable in a format that allows people to search for their intuitions
Anyone who cares about this should friend/follow https://blog.singularitynet.io/the-impact-of-extracellular-matrix-proteins-cross-linking-on-the-aging-process-b7553d375744
Replies from: johnswentworth↑ comment by johnswentworth · 2020-05-14T15:46:17.009Z · LW(p) · GW(p)
Part of the point here is that the models should be firmly nailed down. The field moves fast in large part because old (and many current) models/theories were quite speculative; they were not nailed down.
This would not be a tool for model-making (like a database would be). It would just be a model, along with the evidence/math to validate each component of that model, end-to-end.
That said, sure, a linear presentation is not great.
Replies from: alex-k-chen, alex-k-chen↑ comment by Alex K. Chen (parrot) (alex-k-chen) · 2020-05-14T22:38:05.594Z · LW(p) · GW(p)
IDEALLY, such a model would allow people to creative putative links to hand-annotate (with a dropdown menu) all the papers in support and against support of the model. https://genomics.senescence.info/genes/ exists but it isn't great for mechanism as it's just a long list of genes that seems to have been insight-free scraped. A lot of the aging-related genes people have studied in-depth that have shown the strongest associations for healthy aging (eg foxo3a/IGF1) sure *help* and then there are IGF mutants [oftentimes they dont directly increase repair] but I don't feel that they're as *fundamental* as, say, variations in proteasome function or catalase or splicesome/cell cycle checkpoint/DNA repair genes.
Replies from: ChristianKl↑ comment by ChristianKl · 2020-05-15T12:33:57.547Z · LW(p) · GW(p)
I think you could enter such information into Wikidata.
↑ comment by Alex K. Chen (parrot) (alex-k-chen) · 2020-05-14T22:17:48.387Z · LW(p) · GW(p)
Ok so here's a model I'm thinking. Let's focus on the proteasome alone for instance, which basically recycles proteins. It pulls a protein through the 19S subunit into the 20S barrel that has the "fingers" that can deaminate the amino acids of each protein, one by one.
We know that reduction in proteasome function is one of the factors associated with aging, esp b/c damage in proteasome function accelerates the damage of *all other proteins* [INCLUDING transcription factors/control factors for ALL the other genes of the organism] so it acts as an important CONTROL POINT in our complex system (we also know that proteasome function declines with age). We also know that increases in certain beta3/https://en.wikipedia.org/wiki/PSMB3 subunits of 20S proteasome function help increase lifespan/healthspan (ASK THE QUESTION THEN: why uniquely beta3 more so than the other elements of the proteasome?). Proteins only work in complexes, and this often requires a precise stoichiometry of proteins in order to fit - otherwise you may have too much of one protein in a complex, which [may or may not] cause issues. Perhaps the existence of some subunits help recruit some *other* subunits that are complementary, while it may do negative interference on its own synthesis [there's often an upstream factor telling the cell not to synthesize more of a protein].
I know one prof studies Rpn13 in particular.
The proteasome has a 20S and two 19S regulatory subunits. The 19S subunit consists of 19 individual proteins. Disruptions in synthesizing *any* of the subunits or any of the proteins could make the protein synthesis go wrong.
We need to know:
- is reduction in proteasome function primarily due to reduced proteasome synthesis [either through reduced transcription, splicosome errors, reduced translation, improper stoichiometry, or mislocalization] or damaged proteasomes that continue to stay in the cell and wreak havoc?
- Can proteasomes recognize and degrade proteins with amino acids that have common sites of damage? (many of them known as non-enzymatic modifications)?
- the pdb parameters of proteasomes (as well as the rough turnover rates of each of their subunits)
- what are the active sites of proteasomes, and what amino acids do they primarily consist of? (in particular, do they consist of easily damaged amino acids like cysteines or lysines?)
- What are the precise mechanisms in which the active sites of proteasomes get damaged?
- How does a cell "clear out" damaged proteasomes? What happens to damaged proteasomes during mitosis?
- If a cell accumulates damaged proteasomes, how much do these damaged proteasomes reduce the synthesis and function of other properlyly functioning proteasomes in the cell. Will the ubiquitin system improperly target some proteins into proteasomes that have ceased to exist?
↑ comment by Alex K. Chen (parrot) (alex-k-chen) · 2020-05-14T22:24:05.874Z · LW(p) · GW(p)
Each protein also has to be analyzed in and of itself, b/c upstream of each protein contains numerous alternative splicing variants, and proteins with more splicing variants should presumably be more susceptible to mis-translation than proteins with fewer splicing variants [splicesome function also decreases with age - see william mair on this, so we need a whole discussion on splicesomes, especially as to how they're important for important protein complexes on the ETC].
Proteins also have different variants between species (eg bowhead whales and kakapos have hypofunctioning p53). They have different half-lives in the cell - some of them have rapid turnover, and some of them (especially the neuronal proteins are extremely long-lived proteins). The extremely long-lived proteins (like nuclear pore complexes or others at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5500981/ ) do not go through "degradation/recycling" as frequently as short-lived proteins, so it may be that their rate of damage is not necessarily reduced AS MUCH by increases in autophagy [THIS HAS TO BE MAPPED - there is a lot of waste that continues to accumulate in the cell when it can't be dumped out into the bloodstream/kidneys, and glomeular filtration rate declines with age].
We have to map out which proteins are **CONTROLLERS** of the aging rate, such as protein-repair enzymes [ https://www.sciencedirect.com/science/article/abs/pii/S0098299712001276 ], DNA damage sensing/repair enzymes, Nrf2/antioxidant response elements, and stabilizing proteins like histones [loss of the histone subunits often accelerates aging of the genome by exposing more DNA as unstable euchromatin where it is in more positions to be damaged]. [note i dont include mTOR complex here b/c mTOR reduction is easy but also b/c mTOR doesn't inherently *damage* the cell]
Replies from: johnswentworth, alex-k-chen↑ comment by johnswentworth · 2020-05-15T16:00:45.454Z · LW(p) · GW(p)
That article listing long-lived proteins is a handy one. I'm highly suspicious of a lot of those; radioisotope methods are gold-standard but a large chunk of the listed results are based on racemization and other chemical characteristics, which I don't trust nearly as much. That lack of trust is based more on priors than deep knowledge at this point, though; I'll have to dig more into it in the future.
I don't think "controllers of the aging rate" are quite the right place to focus; there's too many of them. The things I've been calling "root causes" should be less numerous, and "controllers of the aging rate" would be exactly the things which are upstream of those root causes - i.e. things which cause the root causes to accumulate faster/slower over the course of life. (Side note: I think using the phrase "root cause" has been throwing a lot of people off; I'm considering switching to "mediator of history", i.e. things which mediate the effect of the aged organism's history on its current state.)
Replies from: alex-k-chen↑ comment by Alex K. Chen (parrot) (alex-k-chen) · 2020-05-16T02:17:07.241Z · LW(p) · GW(p)
If you had the perfect bioinformatics database + genomically-obsessed autist, it would be easier to deal with larger quantities of genes. Like, the human genome has 20k genes, and let's say 1% are super-relevant for aging or brain preservation - that would be 2k genes, and that would be super-easy for an autistically-obsessed person to manage
Replies from: dogiv, johnswentworth, alex-k-chen↑ comment by johnswentworth · 2020-05-16T02:56:12.629Z · LW(p) · GW(p)
I mean, sure, if we had a really fast car we could drive from New York to Orlando by going through Seattle. But (a) we don't have that amazing database, and (b) it's probably easier to be more efficient than to build the perfect bioinformatics database. With a focus on very-slow-turnover factors, the problem is unlikely to involve even 200 genes, let alone 2k.
You personally might very well be able to identify the full list of root causes, to a reasonably-high degree of certainty, without any tools beyond what you have now, by being more strategic - focusing effort on exactly the questions which matter.